




Quando um medicamento falha por contaminação microbiana, o risco não fica no lote, chega ao paciente. É para impedir essa passagem que o laboratório de microbiologia atua como barreira protetora no controle de qualidade industrial, monitorando matérias-primas, água, ambientes, equipamentos e operadores, além de validar ensaios críticos como o Teste de Endotoxinas Bacterianas (BET), que exige comprovação de sensibilidade, ausência de interferências e critérios rigorosos de aceitação. A avaliação é da diretora técnica e farmacêutica responsável do L.C.Q.Pq. – Laboratório de Controle de Qualidade e Pesquisa Ltda, Marisa de Moura Souza da Luz.
Assim, o laboratório de microbiologia desempenha um papel fundamental no controle de qualidade industrial, especialmente na indústria farmacêutica, ao garantir que os produtos estejam livres de contaminação microbiana.
Ela afirma que a presença de bactérias e fungos (microrganismos aeróbios mesófilos) e microrganismos prejudiciais (patogênicos) pode comprometer a segurança, eficácia e pureza dos medicamentos, representando sérios riscos à saúde dos pacientes. Seu papel inclui:
• Detecção de contaminações em produtos, matérias-primas, ambientes e equipamentos;
• Avaliação da eficácia de processos de esterilização, desinfecção (equipamentos) e sanitização (mãos);
• Monitoramento ambiental em áreas limpas (estéreis) e não estéreis: ar, superfícies planas, côncavo-convexas (equipamentos e bico de enchedoras) e operadores (mãos, jalecos).
Papel do laboratório de microbiologia no controle de qualidade industrial
“O laboratório de microbiologia é responsável por garantir que os produtos, matérias-primas, águas e ambientes estejam microbiologicamente controlados. Ele atua como uma barreira protetora contra a contaminação microbiana”, explica Marisa. Ela acrescenta as seguintes funções:
- Análise de matérias-primas: verifica a ausência ou o nível aceitável de contaminação microbiana nos materiais usados na fabricação.
• Monitoramento ambiental:
- Monitoramento ambiental em áreas não estéreis - avalia a qualidade do ar, de superfícies planas (Placas Rodac), superfícies côncavo-convexas (desinfecção de equipamentos e bico de enchedoras) e operadores (sanitização das mãos).
- Monitoramento ambiental em áreas limpas (estéreis) - avalia a qualidade microbiológica do ambiente de produção, incluindo o ar, superfícies, equipamentos e pessoal, especialmente em áreas de alta assepsia ("salas limpas"). Avaliação da eficácia dos processos de esterilização.
- Testes de bioburden: Bioburden (contagem microbiana e pesquisa de patógenos) - ISO 1173 – 1:2018 - Methods of Determination and Microbial Characterization of Bioburden. USP 44/NF 39 – Vol. 1, 2021. <1115> Bioburden Control of Nonsterile Drug Substances and Products.
• Testes de esterilidade: em medicamentos injetáveis ou estéreis, verifica se o produto final está totalmente livre de microrganismos. Avaliação da eficácia de processos de esterilização, desinfecção (equipamentos) e sanitização (mãos).
• Testes de contagem microbiana: determina a quantidade de microrganismos viáveis em produtos não estéreis para assegurar que os níveis estejam dentro dos limites regulamentados.
• Testes de endotoxinas: detecta a presença de endotoxinas bacterianas, que podem ser tóxicas e causar reações graves em pacientes. USP Chapter <85> Bacterial Endotoxins Test.
• Testes de eficácia de conservantes: avalia se os conservantes em produtos não estéreis são capazes de inibir o crescimento microbiano ao longo do tempo.
• Validação de processos: garante que os processos de limpeza, desinfecção e esterilização são eficazes na eliminação de microrganismos.
Por que é tão crítico para a segurança dos medicamentos
A atuação do laboratório de microbiologia é crítica para a segurança dos medicamentos por várias razões:
Proteção do paciente: a contaminação microbiana em medicamentos pode causar infecções graves e potencialmente fatais, principalmente em pacientes imunocomprometidos ou em produtos injetáveis e oftálmicos.
Prevenção da perda de eficácia: microrganismos podem degradar os ingredientes ativos dos medicamentos, tornando-os menos eficazes ou inativos. A contaminação pode comprometer a estabilidade do produto e sua vida útil.
Conformidade regulatória: a indústria farmacêutica é rigorosamente regulamentada por agências, como a Anvisa. O laboratório de microbiologia deve assegurar que a produção esteja em conformidade com as Boas Práticas de Fabricação (BPF). Seção III - Boas Práticas de Laboratório (BPL) e de Controle de Qualidade (CQ) - Art. 240. O laboratório de microbiologia deve ser organizado de forma a minimizar o risco de contaminação cruzada. (RDC Nº 658/2022 - Anvisa).
Redução de riscos financeiros e de reputação: um lote contaminado pode resultar em recalls de produtos, multas e danos significativos à reputação da empresa. O controle microbiológico eficaz previne esses problemas.
Garantia da qualidade: o laboratório fornece a garantia de que cada lote de medicamento é seguro, puro e eficaz antes de ser liberado para o mercado, protegendo a saúde pública.
Avaliação microbiológica de água purificada e injetável (PW/WFI): é crítico porque contaminações microbianas ou presença de endotoxinas podem causar infecções graves, reações pirogênicas ou comprometer a eficácia do medicamento.
O laboratório atua, portanto, como uma barreira de segurança entre o processo produtivo e o paciente.
Como funcionam os métodos de detecção de endotoxinas
O Teste de Endotoxina Bacteriana é usado para detectar ou quantificar endotoxinas de bactérias gram-negativas presentes em amostras para as quais o teste é preconizado. Utiliza-se o extrato aquoso dos amebócitos circulantes do Limulus polyphemus ou do Tachypleus tridentatus, preparado e caracterizado como reagente LAL.
As endotoxinas bacterianas (lipopolissacarídeos de bactérias gram-negativas) são detectadas por métodos baseados em Lisado de Amebócitos de Limulus (LAL) da Farmacopeia Brasileira 7ª ed., 2024 (FB7) ou Bacterial Endotoxins Test (BET) da Farmacopeia Americana (USP). Ambos são a mesma coisa, BET é a sigla em inglês para Bacterial Endotoxin Test.
Há duas técnicas com sensibilidade diferente para esse teste:
1. Método de Coagulação em Gel (Gel Clot): baseado na formação de coágulo ou gel, indica presença de endotoxina (método semiquantitativo).
2. Métodos Fotométricos quantitativos, que incluem:
- Método turbidimétrico: baseado no desenvolvimento de turbidez após quebra de um substrato endógeno.
- Método cromogênico (cinético ou estático): a endotoxina desencadeia a liberação de um cromóforo, resultando em desenvolvimento de cor após quebra de um complexo peptídico sintético cromogênico.
Receba nossas notícias por e-mail: Cadastre aqui seu endereço eletrônico para receber nossas matérias
Marisa afirma que qualquer um destes procedimentos pode ser realizado, a menos que indicado o contrário na monografia. No método de coagulação em gel, a determinação do ponto final da reação é feita a partir de diluições da substância sob teste em comparação direta com diluições paralelas da endotoxina padrão. As quantidades de endotoxinas são expressas em unidades de endotoxina (UE) definidas. Nota: 1 UE é igual a 1 UI (unidade internacional).
O reagente LAL (lisado de amebócito de Limulus sp.) é preparado para as leituras turbidimétricas ou colorimétricas e estes procedimentos podem ser utilizados se cumprirem os requisitos dos métodos.
“Para sua calibração, é necessária a elaboração de uma curva padrão, obtendo-se a sua regressão linear, na qual se determina, por interpolação, a concentração de endotoxina da substância sob teste”, comenta ela.
Existe um jeito mais simples de colocar os testes de endotoxina em conformidade e melhorar a sustentabilidade com um novo ensaio de endotoxina compendial usando microfluídica. “Esse método simplifica a especificação do teste, diminui as taxas de reteste, cumpre plenamente as diretrizes de integridade de dados mais recentes e reduz o uso de LAL em até 90%”, diz. Para garantir a segurança do paciente, o Capítulo Geral da USP <85> Teste de Endotoxinas Bacterianas (BET) recomenda que a maioria dos medicamentos não deve exceder uma exposição máxima a endotoxina de 5 UE/kg (dentro de 1 hora), considerando um peso médio do paciente de 70 kg.
Desafios da Validação
Os critérios de aceitação da USP <85> para testes de endotoxinas bacterianas envolvem uma validação em várias etapas do próprio teste e o cálculo do limite de endotoxina para o produto específico. O teste é considerado válido se a curva padrão for aceitável (r ≥ 0,980), o controle positivo do produto (Solução B) apresentar uma recuperação de endotoxina entre 50% e 200% e o controle negativo (Solução D) estiver dentro do limite do branco exigido. A aceitação final do produto é determinada por um cálculo que compara a dose máxima do produto com o limiar pirogênico.
“A validação de endotoxina bacteriana é um processo que detecta e quantifica endotoxinas (componentes da parede de bactérias gram-negativas) em produtos como medicamentos injetáveis e dispositivos médicos. Os métodos mais comuns são o LAL, que utiliza reações de coagulação, turbidez ou cor”, ressalta ela.
A validação garante que o teste é adequado para um produto específico e que resultados confiáveis serão obtidos. É a confirmação de que o teste de endotoxina é capaz de analisar com precisão a presença e quantidade de endotoxinas no produto, levando em consideração as características da amostra. É essencial para garantir a segurança do paciente, pois as endotoxinas podem causar febre e resposta inflamatória.
A validação é realizada por meio de teste de inibição ou potencialização descrito para cada uma das técnicas indicadas. São incluídos controles negativos apropriados. A validação deve ser repetida se houver mudança na origem do reagente LAL, no método de produção ou na formulação da substância sob teste.
Testes de interferências no método coagulação em gel (Inibição/Potencialização)
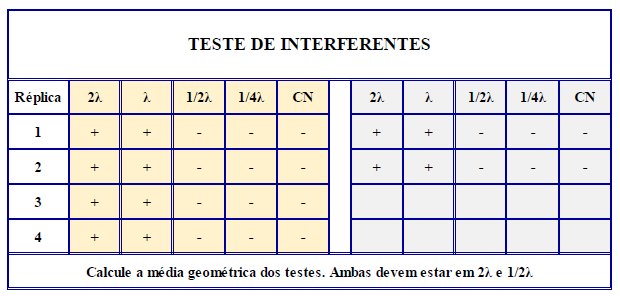
Na prática, se ocorrer a gelificação do 1/4λ significa que algo na amostra está potencializando a reação; se o 2λ não gelificar, significa que algo na amostra está inibindo a reação. O 2λ sempre deve gelificar, porque ele tem o dobro da concentração de endotoxina que o reagente é capaz de detectar (por isso é sempre o controle positivo); e o 1/4λ nunca deverá gelificar, porque já tem 1/4 da concentração de endotoxina que o reagente é capaz de detectar. A média geométrica vai mostrar, numericamente, este resultado: o valor deverá estar entre 2λ e 1/2λ.
No caso de endotoxinas, a validação se chama Teste de Interferências (FB7). Isso porque os produtos farmacêuticos geralmente possuem componentes que acabam por interferir no teste de endotoxinas, então neste teste o que se faz basicamente é: Fazer o teste na amostra na MDV (Máxima Diluição Válida) escolhida para validação, adicionando as concentrações de endotoxina de 2λ, λ, 1/2λ e 1/4λ e na amostra sem adição de endotoxina, tudo isso em quadruplicata.
Em paralelo, faz também duas réplicas de água apirogênica com as concentrações de 2λ, λ, 1/2λ e 1/4λ. O resultado da amostra tem que mostrar que não há interferência do produto no teste, ou seja, tem que oferecer um resultado que esteja entre 2λ e 1/2λ. Feitas as análises, precisa-se calcular a média geométrica (tal qual na curva de confirmação de sensibilidade).
Marisa explica que, na prática, se ocorrer a gelificação do 1/4λ significa que algo na amostra está potencializando a reação; se o 2λ não gelificar, significa que algo na amostra está inibindo a reação. O 2λ sempre deve gelificar, porque ele tem o dobro da concentração de endotoxina que o reagente é capaz de detectar (por isso é sempre o controle positivo); e o 1/4λ nunca deverá gelificar, porque já tem 1/4 da concentração de endotoxina que o reagente é capaz de detectar. A média geométrica vai mostrar, numericamente, este resultado: o valor deverá estar entre 2λ e 1/2λ.
No caso de endotoxinas, a validação se chama teste de interferências. Isso porque os produtos farmacêuticos geralmente possuem componentes que acabam por interferir no teste de endotoxinas, então nesse teste o que se faz basicamente é:
- Amostra MDV em endotoxina refere-se à Máxima Diluição Válida (MDV) para um teste de endotoxinas. É a diluição máxima em que a concentração de endotoxinas de uma amostra pode ser determinada com precisão. Esse valor é crucial para garantir a segurança do paciente, assegurando que, mesmo após a diluição necessária para o teste, a sensibilidade do método ainda seja suficiente para detectar endotoxinas no limite seguro.
- Interferência da amostra: certos excipientes, conservantes ou pH extremos podem inibir ou aumentar a reação.
- Curva padrão e sensibilidade: deve ser validada para cada matriz.
- Ensaios de recuperação: é necessário demonstrar que a recuperação de endotoxina adicionada está entre 50% – 200%. (isso é para técnicas fotométricas (quantitativas). No caso do gel-clot haverá a formação de coágulo, já nas técnicas fotométricas (cromogênicas ou turbidimétricas), o que vai acontecer é que como a concentração de endotoxina é medida, a recuperação da endotoxina adicionada deverá aparecer entre 50% – 200%. Se ficar menos de 50% indica inibição, se ficar mais de 200% indica potencialização.
- Substituição por métodos recombinantes: como rFC (factor C recombinante), que eliminam o uso de animais, exigindo comparabilidade com LAL tradicional.
“A validação do teste de endotoxina bacteriana (BET – Bacterial Endotoxins Test) é uma etapa crítica no controle de qualidade de produtos farmacêuticos, biotecnológicos e dispositivos médicos. Esse teste garante que o produto esteja livre de níveis inaceitáveis de endotoxinas (lipopolissacarídeos de bactérias gram-negativas), que podem causar febre e choque em pacientes”, alerta ela.
Receba nossas notícias por e-mail: Cadastre aqui seu endereço eletrônico para receber nossas matérias diariamente
Desafios na validação do BET
A seguir, Marisa ressalta os principais desafios enfrentados durante a validação do BET (seja pelo método gel-clot, turbidimétrico ou cromogênico):
1. Interferência da matriz do produto
Descrição: componentes do produto (como proteínas, sais, conservantes, surfactantes ou pH extremos) podem inibir ou potencializar a reação entre o reagente LAL (Lisado de Amebócito de Limulus) e as endotoxinas.
Impacto: falsos negativos (inibição) ou falsos positivos (potenciação).
Solução: realizar o teste de inibição/potenciação durante a validação, conforme USP <85> / EP 2.6.14, para comprovar que o método é adequado para o produto específico.
2. Escolha adequada do método
Descrição: existem três principais metodologias — gel-clot, turbidimétrica e cromogênica — com sensibilidades e robustez diferentes.
Impacto: a escolha incorreta pode levar a baixa sensibilidade, falhas na leitura ou incompatibilidade com a amostra.
Solução: selecionar o método com base na natureza do produto, nível de detecção necessário e interferentes conhecidos.
3. Determinação do Limite de Endotoxina (EL)
Descrição: o cálculo incorreto do limite de endotoxina bacteriana (em EU/mL ou EU/mg) pode levar a interpretações erradas dos resultados.
Impacto: aprovação indevida de lotes contaminados ou reprovação injustificada.
Solução: seguir rigorosamente as fórmulas definidas em compêndios oficiais (USP, EP, JP) considerando dose máxima e via de administração.
4. Sensibilidade e controle do reagente LAL
Descrição: o reagente LAL deve ter sensibilidade comprovada e estável (em EU/mL). Lotes diferentes podem apresentar variação.
Impacto: inconsistência nos resultados entre testes ou entre lotes de reagente.
Solução: qualificar cada novo lote e armazenar o LAL conforme as condições recomendadas (temperatura e umidade controladas).
5. Reprodutibilidade e precisão dos resultados
Descrição: variações entre analistas, equipamentos e condições ambientais podem afetar os resultados.
Impacto: dificuldade em comprovar robustez e reprodutibilidade durante a validação.
Solução: implementar treinamento técnico rigoroso, padronizar procedimentos (SOPs) e realizar estudos de precisão inter e intra-analista.
6. Controle da água e do sistema de limpeza
Descrição: a água utilizada (como WFI – Water for Injection) pode ser fonte de endotoxinas se não monitorada.
Impacto: contaminação cruzada ou falsos positivos.
Solução: monitoramento contínuo do sistema de purificação e validação periódica de limpeza e esterilidade.
7. Uso de endotoxina de referência e preparo de diluições
Descrição: as endotoxinas padrão (como a Control Standard Endotoxin) devem ser manuseadas e diluídas corretamente.
Impacto: erros na preparação levam a curvas-padrão incorretas e falhas na quantificação.
Solução: usar diluições seriadas precisas e registrar todos os passos de preparação.
8. Conformidade regulatória e documentação
Descrição: o processo de validação deve seguir normas especificas (USP <85>, EP 2.6.14, JP 4.01, ANVISA RDC 166/2017, etc.).
Impacto: falta de rastreabilidade ou documentação incompleta pode invalidar a validação perante auditorias.
Solução: manter registros completos, protocolos de validação, relatórios e evidências de aceitação dos critérios.
9. Treinamento e qualificação de pessoal
Descrição: o teste BET e sensível a detalhes técnicos (pipetagem, tempo, temperatura).
Impacto: erros humanos podem comprometer toda a validação.
Solução: capacitação continua e qualificação formal de analistas antes de executar ensaios rotineiros.
Como atuar na área
Para se capacitar e estar apto para atuar na indústria farmacêutica, ou mesmo, ascender na carreira, o ICTQ oferece cursos de pós-graduação nessas áreas, por exemplo:
- Gestão da Qualidade e Auditoria na Indústria Farmacêutica (Ao Vivo / On-line) – aborda Boas Práticas de Fabricação de Medicamentos, garantia de qualidade: ferramentas, indicadores e CAPA, documentação técnica aplicada ao sistema de gestão da qualidade, análise e gerenciamento de riscos, revisão periódica de produto, qualificação e validação, integridade de dados etc.
- Assuntos Regulatórios na Indústria Farmacêutica (Ao Vivo / On-line) - voltada aos assuntos regulatórios que regem a indústria farmacêutica no Brasil, farmacovigilância, estudos de equivalência e bioequivalência, vigilância sanitária em portos, aeroportos e fronteiras, legislação tecnovigilância, registro de insumos farmacêuticos, pós-registro de medicamentos, auditoria farmacêutica etc.
- P&D Analítico e Controle de Qualidade na Indústria Farmacêutica – inclui o desenvolvimento para o manejo de técnicas e instrumentação analítica que auxiliarão em competências na pré-formulação e formulação de medicamentos. Além disso, o conteúdo inerente à validação de processos dará maior segurança para atuar na produção e desenvolvimento de medicamentos.
- Gestão e Tecnologia Industrial Farmacêutica - qualificação de fornecedores, boas práticas de fabricação, de laboratório, métodos cromatográficos, validação de processos etc.
Participe também: Grupos de WhatsApp e Telegram para receber notícias



