





A Validação de Limpeza está entre os assuntos mais importantes tratados mundialmente na indústria farmacêutica fabricante de medicamentos e produtos para saúde, através deste estudo é possível avaliarmos os grandes riscos em Contaminação Cruzada que ocorrem entre produtos fabricados e outros agentes físico-químicos e microbiológicos.
A Agência Nacional de Vigilância Sanitária (ANVISA) através das regulamentações sanitárias RDC658/2022 e IN 138/2022, nos trouxe informações importantes que suportam ações a serem realizadas para melhor condução dos nossos estudos, mas, isso não é tudo, outras organizações também tem colaborado e se apresentado como fontes importantes de informação, nos auxiliando a solucionar problemas para a correta aplicação dos conceitos e condução dos estudos, são elas ISPE – International Society for Pharmaceutical Engineering, PDA - Parenteral Drug Association, SINDUSFARMA – Sindicato das Indústrias Farmacêuticas de São Paulo, entre outros.
Neste artigo trataremos da Validação de Limpeza de forma clara e objetiva, trazendo como foco a atualização do tema e desmistificando dúvidas de profissionais do meio farmacêutico.
Receba nossas notícias por e-mail: Cadastre aqui seu endereço eletrônico para receber nossas matérias
Sobre o objetivo da Validação de Limpeza, nada mudou em relação a isso, sempre buscamos comprovar a eficácia dos procedimentos de limpeza utilizados nas linhas de fabricação farmacêutica e tudo que está incorporado a elas, equipamentos, utensílios, superfícies, mangueiras e etc. Vale lembrar que este é o único dentre os estudos de validação que nos permite utilizar o chamado “pior caso ou Worst Case”, onde através da seleção deste “produto pior caso” encontramos o maior risco relacionado pós limpeza, o risco de um possível depósito de residual capaz de contaminar (físico-químico ou microbiológico) o produto fabricado subsequentemente. Sendo assim, se formos capazes de garantir, que após a limpeza deste pior caso todo residual físico-químico ou microbiológico foi removido a níveis seguros, então estaremos seguros de que a limpeza será eficiente para todos os demais produtos contemplados nesta linha.
Para tal avaliação, é imprescindível aplicar o ciclo de vida em Validação de Limpeza, determinado pelas fases 1, 2 e 3, ao qual falaremos logo mais. Em resumo, é necessário elaborar um procedimento de limpeza correto, realizar a seleção do pior caso adequadamente, proceder a elaboração de um protocolo com base na gestão dos riscos, que contemple um plano de amostragem e cálculos adequados dos limites de residuais possíveis. Preferencialmente que possamos incluir os estudos complementares de Tempo de Equipamento Sujo e Tempo de Equipamento Limpo e que tenhamos a visibilidade de Holding Time deste período de avaliação. Isso determinaria seguramente o tempo que esse equipamento/linha poderia permanecer sujo e também o período de validade da limpeza. Após a aplicação do plano de amostragem é obrigatória a comparação dos resultados obtidos, através de procedimentos analíticos validados, frente as especificações dos limites máximos permitidos dos residuais. E finalmente, após a verificação que os resultados estão adequados a estes limites, proceder a elaboração de um relatório com o compilado da execução do estudo e uma clara e objetiva conclusão, assim nenhuma dúvida será gerada em relação a correta condução da avaliação.
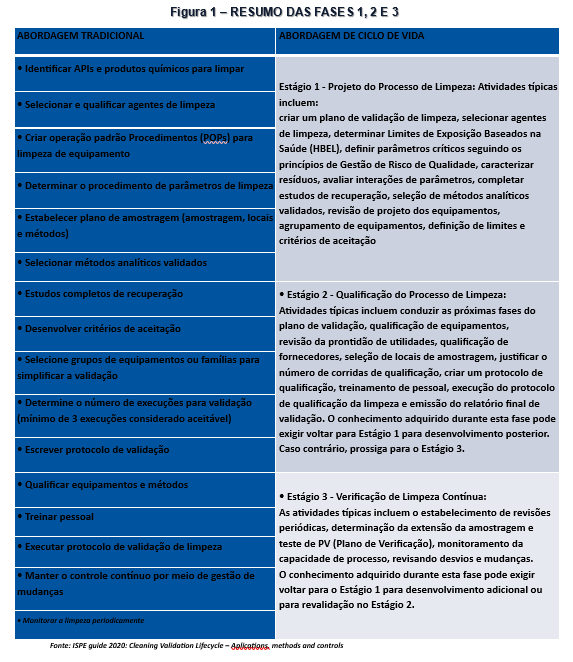
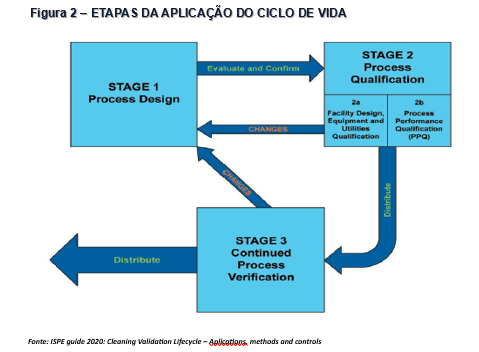
De acordo com o recomendado pela ANVISA e formalizado através da IN 138/2022, sempre que possível, o conceito de ciclo de vida deverá ser aplicado, e este conceito consolidou-se também com a publicação do Guia da ISPE em setembro de 2020 onde todo o documento é embasado em ciclo de vida para a validação de limpeza, o descritivo detalhado das fases está contemplado na sequência abaixo e nas Figuras 1 e 2.
SEQUÊNCIA DE APLICAÇÃO DO CICLO DE VIDA:
FASE 1: Caracterização da Limpeza
- Utilização de Quality by Desing (QbD);
- Caracterização do processo de limpeza em pequena escala para aprimoramento e compreensão do mesmo;
- Avaliação em grande escala;
- Definição do procedimento de limpeza.
- Definição do Pior Caso (Worst Case);
- Definição dos limites de aceitação (realização de cálculos);
FASE 2: Validação da Limpeza
- Elaboração de Protocolo;
- Elaboração Plano de Amostragem com base em riscos
- Execução da Validação;
- Elaboração de Relatório.
FASE 3: Verificação Continuada
- Tratamento estatístico da FASE 2 para definição do Plano de Verificação continuada;
- Definição de limites de alerta;
- Estabelecer testes de monitoramento;
- Definição de pontos mais críticos de amostragem;
- Periodicidade do monitoramento;
- Controle Estatístico do Processo - CEP (controle online);
- Elaboração de Relatório Periódico de Limpeza.
A IMPORTÂNCIA DA AVALIAÇÃO TOXICOLÓGICA NO CENÁRIO DA VALIDAÇÃO DE LIMPEZA:
É importante ressaltar alguns pontos que a regulamentação atual nos trouxe de forma enfática e que sem dúvidas tem sido cobrada com efetividade nas inspeções regulatórias, a avaliação toxicológica das substâncias ativas, também agentes de limpeza e sanitizantes, tal avaliação ocorre basicamente em dois momentos, sobre a seleção do pior caso e sobre a avaliação/cálculos dos residuais. A cobrança da avaliação toxicológica vem na forma dos chamados valores de LEBS - Limite de Exposição com Base em Saúde e PDE/ADE - Exposição Diária Permitida/Aceitável, esses valores são evidenciados por relatórios elaborados por toxicologistas com conhecimento e experiência comprovados.
A avaliação toxicológica é definida como a forma mais segura para determinação dos limites permitidos para limpeza, visando o atendimento do artigo 172 da RDC onde: “um processo de Gerenciamento de Risco da Qualidade, que inclua avaliação toxicológica e de potência, deve ser utilizado para avaliar e controlar os riscos de contaminação cruzada apresentados pelos produtos fabricados”. Com isso, veio a necessidade das empresas realizarem os estudos toxicológicos do portifólio comercial. Pedimos atenção para os prazos, que finalizam em outubro/2023.
UMA NOVA VERTENTE PARA SELEÇÃO DO PIOR CASO:
O cenário atual para seleção do pior caso nos trouxe novidades em relação ao praticado no passado, vale ressaltar que para quem está se atualizando é o momento de incorporar um cenário que é tendência mundial. Para quem já acompanha estudos de validação de limpeza há anos, viu todas as mudanças que a regulamentação proporcionou e percebeu que cada ano que se passa essa seleção fica mais lógica e mais robusta. Nos estudos do passado utilizamos diversos fatores para determinação de um pior caso, como por exemplo: solubilidade do ativo, solubilidade do produto, fator de ocupação, toxicidade pela classe de risco, entre outros, mas, atualmente a recomendação é priorizar a aplicação do critério chamado de Cleanabilidade. O critério através da Cleanabilidade é determinado através da avaliação da solubilidade e concentração de todos os componentes das formulações dos possíveis piores casos de um equipamento/linha, além disso, associa-se a opinião dos colaboradores sobre a dificuldade de remoção do produto durante a limpeza, o que antes já tinha um grande peso e nesta determinação continua sendo um importante fator, porém, tem um peso menor alinhado aos outros fatores anteriormente citados. E o fator que ganhou um peso maior nesta avaliação atualmente, relacionado ao critério Cleanabilidade foi a toxicidade dos IFAs (Insumos farmacêuticos Ativos) das formulações, que será então o fator decisivo para determinação do “worst case”. No caso de empate de produtos durante a seleção do pior caso baseada neste critério, a sequência ser utilizada é toxicidade, solubilidade do ativo em água e dificuldade de remoção.
Em relação aos cálculos, todos eles devem carregam um racional de aplicação as linhas ao qual estão sendo utilizados (dedicados ou multipropósito) e observar se são apropriados ao tipo de residual ao qual se aplicam (microbiológico, ativo, agente de limpeza e/ou sanitizante). Os cálculos para determinação dos limites podem ser realizados através das 3 estratégias existentes: 10ppm, 0,1% da menor dose terapêutica e toxicidade sendo que o valor mais conservador obtido entre estas estratégias deve ser o selecionado para a pesquisa dos resíduos de validação de limpeza.
Esperamos com essas explanações solucionar alguns anseios dos nossos colegas do meio farmacêutico, e ainda provocar a curiosidade nos que não conhecem muito do assunto em aprofundar-se ainda mais no tema. O mercado farmacêutico precisa de profissionais que busquem sempre por novidades e se sigam muito além do que somente sua rotina e seus estudos, precisamos compartilhar aquilo que temos de melhor e multiplicar o que nos fortalece e nos diferencia no mercado farmacêutico, nosso profundo conhecimento sobre as Boas Práticas de Fabricação!
Participe também: Grupo de WhatsApp e Telegram para receber notícias farmacêuticas



