





O objetivo da RDC 73, de 7 de abril de 2016, da Agência Nacional de Vigilância Sanitária (Anvisa), é garantir que um medicamento registrado mantenha as características de qualidade, segurança e eficácia, mesmo após a realização de mudanças.
Em termos gerais, uma mudança pós-registro é qualquer alteração realizada na condição registrada do medicamento que tenha impacto em composição, equipamentos, processo produtivo, controle de qualidade e embalagem, entre outros.
“É muito importante que as indústrias farmacêuticas estejam atualizadas com as novas tecnologias do setor, não somente com relação aos equipamentos, processos, composição e outros, mas também com relação às novas tecnologias de métodos analíticos que são utilizados no controle de qualidade das matérias-primas, insumo farmacêutico ativo (IFA) e do medicamento”, lembra a coordenadora de Assuntos Regulatórios Pós-Registro e professora do ICTQ – Instituto de Pesquisa e Pós-Graduação para o Mercado Farmacêutico, Lilian Agapito.
Ela ressalta que a RDC 73/16 é fundamental na medida em que propõe mais mudanças pós-registro tratadas como de implementação imediata. Essas mudanças não precisam de uma aprovação prévia da Anvisa para sua implementação. Assim, a medida é importante para as indústrias farmacêuticas realizarem a manutenção do portfólio em tempo hábil e, para a Anvisa, diminuir a fila de petições pós-registros que ficam aguardando aprovação.
Outro ponto importante que a professora menciona é o desmembramento de mudanças previstas na RDC 48/09 e a classificação de mudanças que antigamente não estavam previstas nesta norma.
“Por exemplo, a RDC 73/16 traz as mudanças de especificações e métodos analíticos para IFA e para excipientes também, enquanto que a RDC 48/09 tratava apenas de produto acabado. Além disso, a RDC 73/16 traz oito mudanças para especificações e métodos, enquanto que a RDC 48/09 trazia apenas dois tipos de mudança”, destaca ela.
Para Lilian, esta RDC traz a mudança de substituição ou inclusão de local de fabricação do IFA do mesmo grupo farmoquímico, que é de implementação imediata após protocolo na Anvisa. Enquanto que para a RDC 48/09, esse tipo de mudança aguardava deferimento.
“O último ponto a citar aqui é o Parecer de Análise Técnica da Empresa (PATE). Este documento visa auxiliar que a mudança proposta foi avaliada criteriosamente, com análise do seu potencial impacto na qualidade, segurança e eficácia do medicamento”, destaca ela.
Segue abaixo a RDC 73/16, com todos os seus artigos na íntegra. Para facilitar o entendimento do leitor, nós estamos dispondo todas as análises e interpretações no decorrer do texto da norma, em negrito, logo após cada artigo em questão. Além disso, como os anexos são muito extensos, apenas seus títulos estão sendo citados no final da RDC. Os comentários, tanto nos artigos como nos anexos, foram feitos pela professora Lilian.
Resolução RDC n° 73, de 7 de abril de 2016
Dispõe sobre mudanças pós-registro, cancelamento de registro de medicamentos com princípios ativos sintéticos e semissintéticos e dá outras providências.
A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe conferem o art. 15, III e IV aliado ao art. 7º, III, e IV, da Lei nº 9.782, de 26 de janeiro de 1999, o art. 53, V, §§ 1º e 3º do Regimento Interno aprovado nos termos do Anexo I da Resolução da Diretoria Colegiada - RDC n° 61, de 3 de fevereiro de 2016, resolve adotar a seguinte Resolução da Diretoria Colegiada, conforme deliberado em reunião realizada em 22 de março de 2016, e eu, Diretor-Presidente, determino a sua publicação.
Art. 1º Fica aprovado o Regulamento Técnico que estabelece os procedimentos para mudanças pós-registro e cancelamento de registro de medicamentos, nos termos desta Resolução.
CAPÍTULO I
DAS DISPOSIÇÕES INICIAIS
Seção I
Objetivo
Art. 2º Esta Resolução tem o objetivo de classificar as mudanças pós-registro de medicamentos, estabelecer os critérios e a documentação mínima necessária, prever responsabilidades diretas das empresas e estabelecer o procedimento simplificado de mudanças pós-registro de implementação imediata de acordo com a classificação da mudança estabelecida neste regulamento, visando garantir a qualidade, segurança e eficácia destes medicamentos.
Quando se fala em prever responsabilidades diretas das empresas, este é um dos grandes diferenciais desta norma em detrimento das resoluções pós-registro. Não que as normas anteriores não cobravam isso, a responsabilidade da empresa frente às mudanças sempre existiu, porém nesta norma há algumas mudanças tais como, assinaturas de documentos técnicos em que há assinatura não só pelo responsável técnico, mas também pelo responsável da Garantia da Qualidade, Assuntos Regulatórios e no PATE. Além destes, é necessária também a assinatura pelo departamento responsável pela mudança.
Seção II
Abrangência
Art. 3º Esta Resolução se aplica a todos os medicamentos com princípios ativos sintéticos e semissintéticos classificados como novos, similares e genéricos.
A RDC 48/09 se aplicava também para medicamentos específicos. Atualmente as mudanças pós-registro para medicamentos específicos está regulamentada pela RDC 76/16.
Seção III
Definições
Art. 4º Para efeito desta Resolução, são adotadas as seguintes definições:
I - Histórico de Mudanças do Produto (HMP): documento disponível na empresa no qual deverão ser registradas informações a respeito do histórico anual do produto;
II - Protocolo de estudo de estabilidade: documento por meio do qual se define o plano de estudo de estabilidade, incluindo as provas e critérios de aceitação, cronograma, características do lote a ser submetido ao estudo, quantidade das amostras, condições do estudo, métodos analíticos e material de acondicionamento;
III - Mudanças múltiplas concomitantes: mudanças decorrentes de uma mudança principal prevista nesta Resolução;
IV - Mudanças múltiplas paralelas: duas ou mais mudanças simultâneas e diretamente relacionadas protocoladas conjuntamente;
V - Procedimento ordinário: é o procedimento de peticionamento que requer protocolo e que deve aguardar manifestação favorável da Anvisa para a implementação;
VI - Procedimento simplificado: é a simplificação do procedimento ordinário de peticionamento, exclusivamente para as petições que são classificadas como de implementação imediata por este regulamento;
VII - Parecer de Análise Técnica da Empresa (PATE): parecer elaborado pela empresa detentora do registro que aborda no mínimo todos os critérios e documentos previstos neste regulamento e normativas sanitária afins, incluindo uma avaliação crítica de todos os aspectos relevantes para a avaliação da Anvisa. O mesmo deve assegurar que foram realizados e aprovados os critérios e documentos apresentados para a autoridade sanitária com a finalidade de manutenção dos parâmetros de qualidade, segurança e eficácia do produto;
VIII - Suspensão do Procedimento simplificado: condição na qual a empresa fica impossibilitada de realizar o procedimento simplificado por um determinado período; e
IX - Mudança de implementação imediata: mudança pós-registro para qual a Anvisa concede autorização prévia para sua imediata implementação pela empresa, mediante a inclusão no HMP ou na petição protocolada individualmente, de todas as provas satisfatórias requeridas para a modificação, conforme disposto neste regulamento.
Das definições, o que temos de novidade é o PATE. Este é um documento que surge com a RDC 73/16. É um parecer que aborda vários itens a serem avaliados e discutidos criticamente pela empresa. Atualmente há um Manual da GGMED/Anvisa disponível no site da Anvisa, contendo um modelo de PATE e as orientações para preenchimento. Este manual não é mandatório, é apenas uma recomendação da Anvisa.
Além disso, neste artigo que versa sobre definições temos a definição do item VIII para a Suspensão do Procedimento Simplificado. Esta suspensão será aplicada para empresas que descumprirem com a norma, no caso, a punição será a suspensão do procedimento simplificando, ficando a empresa um ano sem poder realizar mudanças de implementação imediata para todos os produtos de seu portfólio. Esta punição não estava prevista pela norma antiga, RDC 48/09.
CAPÍTULO II
DAS DISPOSIÇÕES SOBRE CLASSIFICAÇÃO E PROTOCOLO DAS MUDANÇAS PÓS-REGISTRO
Art. 5º As mudanças pós-registro são classificadas de acordo com o seu potencial impacto na qualidade, segurança e eficácia do medicamento, podendo ser de implementação imediata, com ou sem protocolo individual, ou depender de aprovação prévia da Anvisa.
§ 1º As mudanças classificadas como de implementação imediata por esta norma, cuja empresa identifique potencial impacto significativo na qualidade, segurança e eficácia do medicamento, deverão ser peticionadas segundo o procedimento ordinário, com assunto pertinente, e aguardarão manifestação da Anvisa para a sua implementação.
§ 2º A empresa suspensa de protocolar segundo o procedimento simplificado, nos termos dos Artigos 36 e 45, deverá protocolar de acordo com o procedimento ordinário todas as mudanças pós-registro de sua titularidade.
As empresas devem realizar análises de riscos para fins de avaliação do impacto da mudança no produto com relação à sua qualidade, segurança e eficácia.
Se, por meio da análise de risco, a empresa identificar que a mudança causa alto impacto na qualidade, segurança ou eficácia do produto, esta mudança deverá seguir o procedimento ordinário, ou seja, protocolar a solicitação de mudança e aguardar o deferimento da Anvisa. Somente após o deferimento a mudança poderá ser implementada.
Conforme comentado no artigo das definições, este artigo traz um parágrafo para versar sobre a parte punitiva da legislação, ou seja, se a empresa tiver sido suspensa de realizar o procedimento simplificado (implementações imediatas), todas as mudanças pós-registros da empresa deverão seguir o procedimento ordinário, e isto se aplica a todo o portfólio.
Art. 6º As mudanças de implementação imediata serão permitidas quando todas as provas requeridas estiverem anexadas ao HMP disponível na empresa ou à petição individual protocolada, exceto quando a referida mudança for paralela a outra que requeira prévia aprovação, hipótese em que a implementação das mudanças e o preenchimento do HMP deverá ser feito somente após a aprovação da Anvisa.
§ 1º A implementação imediata das mudanças não impede a análise, a qualquer tempo, da documentação exigida, podendo ser ratificada ou indeferida.
§ 2º Em caso de indeferimento, as condições anteriores à mudança deverão ser restabelecidas imediatamente após a manifestação da Anvisa ou a fabricação do medicamento deverá ser temporariamente descontinuada.
Este artigo traz claramente quando se dá o momento de uma mudança de implementação imediata. Ressalto aqui que a data da implementação de uma mudança de imediata via HMP não é a data de submissão do HMP no sistema de peticionamento eletrônico da Anvisa, como muitos me questionam, e sim a data em que todas as provas requeridas estão anexadas ao HMP a qualquer momento.
Utilizamos a expressão de HMP vivo, o histórico de mudanças do produto é diário conforme as mudanças vão acontecendo, portanto se a submissão do HMP é em dezembro de 2018, e a mudança ocorreu em junho do mesmo ano, se todas as provas estiverem no HMP da empresa em junho, esta será a data de implementação. Lembrando que, em uma eventual inspeção in loco pelas autoridades sanitárias, caso seja solicitado o HMP de determinado produto, este deve estar integralmente disponível na empresa.
Mesmo se tratando de HMP ou protocolo de mudanças de implementação imediata, a Anvisa pode analisar os documentos a qualquer momento e manifestar um parecer. Desta forma se a Anvisa indeferir e solicitar o retorno à condição registrada e a empresa não estiver preparada para retornar à esta condição, a empresa deverá descontinuar a produção do medicamento alvo do indeferimento.
Um exemplo seriam as mudanças relacionadas às trocas de equipamentos. Imaginem uma troca de equipamentos em que os equipamentos obsoletos não estão mais presentes na empresa. Nesta situação, não haveria viabilidade para este retorno acontecer.
Lembrem-se de que estamos falando de várias situações, pois o indeferimento pode ser por vários motivos, porém se este levar à punição prevista nos artigos 36 e 45 desta norma, a empresa não poderá realizar nenhuma mudança de implementação imediata, portanto, ou os equipamentos da condição anteriormente registrada estão disponíveis na empresa para viabilizar o retorno ou a empresa caminhará para uma descontinuação temporária do produto. Nem preciso dizer que este desfecho seria o pior cenário de todos.
Portanto, é importante ressaltar que as avaliações das mudanças devem ser muito criteriosas, tecnicamente fundamentadas para que os enquadramentos sejam precisos.
Art. 7º As mudanças que requeiram aprovação prévia devem ser protocoladas e aguardar análise e manifestação favorável da Anvisa para serem implementadas.
§ 1° após a aprovação a empresa terá até 180 (cento e oitenta) dias para implementação da modificação, exceto quando houver manifestação contrária da Anvisa.
§ 2° Após a produção do primeiro lote com a mudança aprovada, não será permitida a produção de lotes em condição diferente.
Diferentemente da norma anterior RDC 48/09, a RDC 73/16 informa o prazo em que a mudança deve ser implementada, no caso 180 dias. Cumpre esclarecer que este prazo se aplica no caso de mudanças que caracterizam alteração ou substituição, para uma inclusão de local de fabricação do IFA, por exemplo, este prazo não é aplicável, já que a condição registrada anteriormente continua aprovada.
Art. 8º Quando houver mais de uma mudança simultânea para uma mesma apresentação, concentração e forma farmacêutica, a empresa poderá protocolar essas mudanças paralelamente ou concomitantemente.
Este conceito de mudanças paralelas e concomitantes já era definido pela RDC 48/09. Lembrando que mudanças paralelas, salvo exceções de mudanças exclusivas de HMP, há o pagamento das taxas de cada mudança e estas são informadas nos formulários de petição (FP2).
Art. 9º Nos casos de mudanças paralelas, a empresa deverá protocolar cada mudança individual apresentando documentação única que contemple todas as provas relativas a cada um dos assuntos de petição.
§ 1º A descrição das alterações paralelas e sua correlação devem constar na justificativa a que se refere o artigo 15, inciso III, desta Resolução.
§ 2º A requerente deve apresentar a avaliação do efeito aditivo de mudanças individuais paralelas no que se refere ao potencial impacto na qualidade, segurança e eficácia do medicamento e apresentar as provas adicionais, quando necessário.
Importante esclarecer, que várias mudanças menores podem caracterizar em mudança maior devido ao potencial impacto na qualidade, segurança e eficácia do medicamento, por isso é solicitada esta avaliação do efeito aditivo.
Art. 10. Nos casos de mudanças concomitantes, o peticionamento deve ser referente à mudança principal e a informação sobre a mudança concomitante deve ser descrita na justificativa.
§ 1º As únicas mudanças que serão consideradas como concomitantes são aquelas explícitas nesta norma.
§ 2º Devem ser apresentadas as provas relativas a todas as mudanças.
§ 3º Quando a documentação solicitada em mudanças concomitantes for divergente, deverá ser apresentada a documentação relativa à mudança principal.
Para mudanças concomitantes não há a necessidade de pagamentos de taxas, e estas não são informadas nos formulários de petição (FP2), porém, elas devem estar devidamente justificadas e seus documentos previstos nesta resolução devem ser apresentados.
Art. 11. Nos casos das alterações pós-registro não previstas nesta Resolução, a empresa deverá entrar em contato com a Anvisa para estabelecer os testes e a documentação que deverão ser apresentados.
Neste artigo fica oficializado que nenhuma mudança não prevista na norma deva ser realizada sem a devida análise prévia da Anvisa para os possíveis enquadramentos. Portanto, para as mudanças não previstas aqui não hesitem em tratar o assunto com a Anvisa, o que pode ocorrer via Fale Conosco, no site da Anvisa, ou em agendamentos de reuniões no parlatório da Agência.
Art. 12. As mudanças pós-registro previstas nesta Resolução estão descritas no anexo I deste regulamento.
§ 1º As mudanças relacionadas ao insumo farmacêutico ativo estão descritas no anexo I, item 1 (um), modificações a; b; c; d; e.
§ 2º As mudanças relacionadas aos testes, limites de especificações e métodos analíticos do controle de qualidade e estabilidade do insumo farmacêutico ativo e medicamento estão descritas no anexo I, item 2 (dois), modificações a; b; c; d; e; f; g; h.
§ 3º As mudanças relacionadas aos testes, limites de especificações e métodos do controle de qualidade do excipiente estão descritas no anexo I, item 3 (três), modificação a.
§ 4º As mudanças relacionadas a descrição e composição do medicamento estão descritas no anexo I, item 4 (quatro), modificações a; b; c; d; e; f; g; h; i; j; k; l; m; n.
§ 5º As mudanças relacionadas ao local de uma ou mais etapas do processo produtivo do medicamento estão descritas no anexo I, item 5 (cinco), modificações a; b; c; d; e; f; g; h.
§ 6º As mudanças relacionadas ao processo de produção do medicamento, equipamento e tamanho de lote estão descritas no anexo I, item 6 (seis), modificações a; b; c; d; e; f; g.
§ 7º As mudanças relacionadas à embalagem do medicamento estão descritas no anexo I, item 7 (sete), modificações a; b; c; d; e; f; g; h; i; j; k; l.
§ 8º A mudança relacionada a inclusão de nova apresentação está descrita no anexo I, item 8 (oito), modificação a.
§ 9º As mudanças relacionadas ao prazo de validade ou aos cuidados de conservação do medicamento estão descritas no anexo I, item 9 (nove), modificações a; b; c; d.
§ 10º A inclusão de nova concentração estão descritas no anexo I, item 10 (dez), modificações a; b.
§ 11º As mudanças relacionadas à posologia, ampliação de uso, inclusão de nova via de administração e nova indicação terapêutica estão descritas no anexo I, item 11 (onze), modificações a; b; c; d.
§ 12º As mudanças relacionadas ao nome do medicamento, cancelamento do registro do medicamento e exclusão de local de fabricação do fármaco, local de embalagem primária local de embalagem secundária e/ou local de fabricação do produto estão descritas no anexo I, item 12 (doze), modificações a; b; c; d.
Aqui são listadas todas as mudanças previstas nesta norma. Vamos discuti-las item a item no Anexo I.
CAPÍTULO III
DAS DISPOSIÇÕES GERAIS REFERENTES À DOCUMENTAÇÃO
Art. 13. A documentação solicitada para cada modificação está descrita no Anexo I deste regulamento.
Parágrafo único. Quando algum dos documentos exigidos não for aplicável, a não apresentação do mesmo deve ser acompanhada de justificativa técnica e dados que suportem a sua ausência.
A Anvisa atualmente trabalha com um sistema de triagem antes de iniciar a análise técnica da petição. Portanto, se qualquer documento previsto em checklist da mudança não estiver presente ou com sua ausência justificada, ocorrerá um indeferimento sumário da petição, termo utilizado para petições que são indeferidas sem questionamentos (exigências) da Anvisa.
Art. 14. Toda a documentação deve estar de acordo com legislação específica e, existindo guia, este deverá ser consultado e adotado conforme aplicação.
§ 1º Normas específicas, tais como as que estabelecem os critérios de bioisenção, validação de metodologia analítica e estudo de estabilidade, podem servir de fundamento para a ausência de documentação exigida nesta Resolução.
§ 2º Na ausência de legislação e guias específicos, a empresa deverá consultar a Anvisa, previamente à submissão da petição, a apresentação de provas adicionais.
Atualmente existem normas específicas que subsidiam a ausência de determinado estudo, ou que exigem estudos adicionais. Tudo deve ser levado em consideração para justificar ausências de documentos ou a apresentação de documentos adicionais.
Art. 15. Todas as petições de mudanças pós-registro e cancelamento de registro de medicamentos devem ser acompanhadas dos seguintes documentos:
I - Guia de Recolhimento da União relativa à Taxa de Fiscalização de Vigilância Sanitária (TFVS) acompanhada do respectivo comprovante de pagamento ou GRU isenta, quando for o caso;
II - Formulários de Petição devidamente preenchidos;
III - Justificativa da solicitação, contemplando a descrição detalhada e o racional da proposta, conforme Anexo II; e
IV - Parecer de Análise Técnica da Empresa (PATE).
§ 1º A petição do Histórico de Mudanças do Produto dispensa a apresentação de Formulários de Petição.
§ 2º As petições de cancelamento de registro do medicamento e da apresentação dispensam a apresentação do PATE.
§ 3º O solicitante da mudança pós-registro deverá apresentar o PATE em via impressa e em mídia eletrônica, de modo a permitir a realização de busca textual e cópia.
§ 4º O PATE deve ser assinado pelo responsável técnico, responsável pela garantia da qualidade, responsável pelo regulatório da empresa detentora do registro e pelos demais responsáveis pela mudança. Orientações adicionais a respeito do PATE serão disponibilizadas no sítio eletrônico da Anvisa.
Mesmo que o item do Anexo I não cite estes documentos como sendo do checklist da mudança, estes deverão ser apresentados. Atenção para a mídia eletrônica do PATE, de modo a permitir a realização de busca textual e cópia, pois este item é novidade nesta norma pós-registro.
Art. 16. Os dados do estudo de estabilidade gerados posteriormente à apresentação do protocolo de estabilidade ou do estudo de estabilidade incompleto, relativos às petições de implementação imediata e às petições deferidas, deverão ser incluídos no HMP, mesmo que o estudo não esteja concluído.
Trazendo um pouco do histórico deste documento na área regulatória, antigamente, antes da RDC 48/09, não tínhamos o HMP. As petições que aguardavam análise da Anvisa e continham estudos de estabilidades parciais eram atualizados via Aditamentos. Então, era um prática comum realizar aditamentos para enviar informações para a Anvisa dos tempos já analisados dos estudos de estabilidade.
Com a RDC 48/09 surge o HMP. A partir deste momento a regra era não realizar mais os aditamentos, mas informar estes estudos completos no HMP para petições deferidas e para mudanças exclusivas de HMPs, as estabilidades deveriam ser apresentadas mesmo que parciais, isto tudo conforme uma Orientação de Serviço nº 09/GGMED/Anvisa, de 11 de novembro de 2013.
Portanto, estas orientações passam a ser regras regulamentadas por esta norma pós-registro.
Art. 17. Nos casos em que for exigido protocolo de validação de processo, o relatório sumário de validação gerado posteriormente deverá ser incluído no HMP.
Como será observado em algumas mudanças pós-registro, a validação de processo é um item de checklist e compõe o dossiê que será submetido na Anvisa. A validação de processo não era requisitada pela RDC 48/09 para submissão das mudanças, porém a RDC 17/10 de Boas Práticas de Fabricação de Medicamentos já exigia estes estudos que ficam disponíveis na empresa.
Art. 18. Resultados fora de especificação do estudo de estabilidade em andamento devem ser informados imediatamente à Anvisa após investigação preliminar, incluindo a avaliação da necessidade de aplicação de medida cautelar.
Parágrafo único. A proposta de ação corretiva deverá ser enviada posteriormente à conclusão da investigação.
Art. 19. O prazo de validade do medicamento será definido de acordo com os resultados de estabilidade apresentados.
§ 1º Para petições que devem aguardar a manifestação favorável da Anvisa, em que o estudo de estabilidade enviado comprovar prazo de validade provisório inferior àquele registrado, este será reduzido e não será necessário o peticionamento da redução do prazo de validade.
§ 2º Para as petições de implementação imediata, em que o estudo de estabilidade enviado comprovar prazo de validade provisório inferior àquele registrado, a empresa deve peticionar a redução do prazo de validade.
§ 3º Nos casos em que for exigido protocolo de estudo de estabilidade, o prazo de validade registrado será mantido.
Aqui cabe ressaltar que se o estudo de estabilidade apresentado for estudo acelerado completos, ou seja, 6 meses e, estudos de estabilidade de longa duração com resultados parciais de 6 meses, o prazo de validade provisório será de 24 meses.
Art. 20. Os formulários contidos nos Anexos II e IV referidos nesta norma devem ser apresentados de acordo com os modelos propostos.
Parágrafo único. O formulário do anexo II deve ser devidamente assinado pelo responsável técnico, responsável pela garantia da qualidade e responsável pelo regulatório da empresa detentora do registro.
Pela RDC 48/09, a justificativa técnica era assinada somente pelo responsável técnico da empresa. Agora, pela RDC 73/16 devem assinar o responsável técnico, responsável pela garantia da qualidade e responsável pelo regulatório da empresa detentora do registro.
Art. 21. Não será necessário anexar à petição os novos modelos de texto de bula e rotulagem para as alterações pós-registro que necessitem de atualização destes, exceto quando solicitados nesta norma ou a critério da Anvisa.
Parágrafo único. A empresa deverá atualizar as informações na bula e rotulagem de acordo com as mudanças pós-registro.
A atualização da bula segue as regras dispostas pela RDC 60/14 e a notificação de alteração de rotulagem em decorrência de aprovação pós-registro, segue a RDC 61/12.
Art. 22. Nos casos em que a mudança pós-registro se referir a mais de uma concentração de uma mesma forma farmacêutica, esta deverá ser protocolada com ordem de produção de lotes no mínimo referente à maior e menor concentração, desde que as formulações sejam qualitativamente iguais, proporcionais e fabricadas no mesmo local, com o mesmo processo produtivo.
Parágrafo único. Nos casos a que se refere o caput, deve ser apresentada justificativa baseada na comparação das características das formulações e processo produtivo das diferentes concentrações.
Este artigo vem para facilitar a composição dos dossiês, pois no caso de produtos com várias concentrações de uma mesma forma farmacêutica, um dossiê composto com ordem de produção de lotes referente à maior e à menor concentração deixa o processo com um número páginas menor, porém contendo todas as informações técnicas relevantes para a avaliação da mudança.
Art. 23. Nos casos em que sejam propostos mais de um local de fabricação de medicamento, mais de um local de fabricação de fármaco, mais de um processo produtivo ou mais de uma forma de acondicionamento, entre outras alterações, a não apresentação das provas requeridas contemplando todas as combinações possíveis entre as condições registradas e as alterações propostas deve ser fundamentada tecnicamente, com informações e histórico que possam justificar sua ausência.
Este artigo solicita que todas as combinações da condição registrada do medicamento sejam consideradas na mudança e que as ausências de determinadas provas sejam justificadas, como perfil de dissolução comparativo por exemplo. No PATE há um item específico apenas para discutir estas combinações.
Art. 24. Quando uma mudança pós-registro exigir documentos técnicos, como relatório de produção, estudos de estabilidade, laudos de controle de qualidade, entre outros, haverá avaliação em relação às condições de Boas Práticas de Fabricação da empresa fabricante do medicamento existentes no momento da produção dos lotes, relatórios e respectivas análises que foram submetidos à Anvisa.
Parágrafo único. A avaliação das condições de Boas Práticas de Fabricação de que trata o caput poderá resultar na validação ou invalidação dos documentos apresentados.
Atualmente é a RDC 17, de 16 de abril de 2010, que dispõe sobre as Boas Práticas de Fabricação de Medicamentos, portanto, principalmente em casos de inclusões ou alterações de locais de fabricação, é importante certificar se o local possui CBPF.
Art. 25. Para medicamentos similares e genéricos, nas mudanças pós-registro em que é solicitado relatório técnico de estudo de biodisponibilidade relativa/bioequivalência, o estudo deve ser realizado entre o medicamento proposto e o medicamento de referência.
Sabemos que há casos em que não há um medicamento referência eleito pela Anvisa. Para estes casos, a empresa deverá solicitar a inclusão de um medicamento referência na lista. A Anvisa irá avaliar e incluir o produto indicado na lista. Não havendo medicamentos no mercado para indicação, a empresa deverá discutir o caso com a Anvisa para definir a melhor forma de conduzir os estudos.
CAPÍTULO IV
DO HISTÓRICO DE MUDANÇAS DO PRODUTO
Art. 26. O HMP é de responsabilidade da empresa detentora do registro que deverá preencher e anexar a documentação pertinente para cada processo.
Lembrando que se há terceirização, o responsável pelo HMP sempre será o detentor do registro. Com isso, é fundamental haver um bom alinhamento com os terceiros envolvidos em etapas produtivas do medicamento.
Art. 27. Todas as mudanças pós-registro devem ser registradas no HMP simultaneamente à data de sua implementação e/ou aprovação.
Aqui fica claro aquele conceito que citei no início de HMP vivo. O HMP sempre foi entendido desta forma desde a RDC 48/09, pois o HMP é um documento que deve estar disponível na empresa. Portanto, as mudanças devem ser incluídas no HMP em tempo real em que elas ocorrem.
Art. 28. Quando a mudança for de implementação imediata e não necessitar de protocolo individual, a documentação exigida para cada mudança estabelecida no Anexo I desta Resolução, incluindo o PATE, deve ser anexada ao HMP na data da referida implementação.
Salientando, a implementação da mudança somente poderá ocorrer após todas as provas estiverem anexadas no HMP. O PATE deve ser incluído no HMP para todas as mudanças previstas nesta norma que não tem exceção deste documento. Portanto, pode acontecer do HMP conter vários PATEs referentes a mudanças distintas.
Art. 29. O HMP deve conter as seguintes informações:
I - Todas as mudanças pós-registro de implementação imediata, com ou sem protocolo, bem como as que tiveram aprovação prévia da Anvisa;
II - Informações complementares, incluindo:
a) a lista de lotes fabricados ou importados no ano, destinados exclusivamente à comercialização no mercado brasileiro, incluindo data de fabricação, número e tamanho do lote (massa/volume e unidades farmacotécnicas);
b) última versão do(s) documento(s) contendo testes, limites de especificação e métodos analíticos de controle de qualidade do medicamento, conforme aprovado;
c) relatórios de estudos de estabilidade de acompanhamento concluídos e documentos citados nos artigos 16 e 17; e
d) demais informações que não são caracterizadas como mudanças pós-registro, mas que são atualizações de informações apresentadas no registro.
Com relação ao item II, estas informações complementares não eram solicitadas na vigência da RDC 48/09. Notem que a lista de lotes e as especificações e métodos do produto acabado visam a obter um histórico do produto.
Art. 30. O HMP deve estar atualizado e facilmente disponível na empresa para apresentação à autoridade sanitária quando requerido.
Conforme já mencionei nos artigos anteriores, o HMP é um documento que deve estar disponível na empresa contendo todas as mudanças que ocorreram até aquele momento.
Art. 31. Os dados do HMP deverão ser protocolados anualmente, no mês do aniversário do registro do medicamento, mesmo não havendo nenhuma mudança pós-registro, e deverão ser referentes ao período de 12 (doze) meses anteriores ao seu protocolo.
Parágrafo único. O protocolo do HMP deve ser realizado através do peticionamento eletrônico e selecionada a modalidade de petição eletrônica, não havendo a necessidade de envio da documentação em papel.
Este procedimento é pelo sistema eletrônico de peticionamento no site da Anvisa. Trata-se do envio formal para a Anvisa de todas as mudanças que ocorreram para cada produto.
CAPÍTULO V
DAS DISPOSIÇÕES FINAIS E TRANSITÓRIAS
Art. 32. As decisões da Anvisa quanto à avaliação das solicitações pós-registro serão objeto de publicação no Diário Oficial da União, ou em outro meio de divulgação institucional, quando aplicável.
As publicações de deferimentos ou indeferimentos ocorrem rotineiramente às segundas-feiras por meio de um Suplemento publicado no site da imprensa nacional. No entanto, há exceções, pois podem ocorrer publicações fora deste suplemento e em outros dias da semana. Portanto, é importante a leitura de DOU diariamente.
Art. 33. As orientações da Anvisa para as mudanças pós-registro de medicamentos serão disponibilizadas para consulta no site desta Agência.
A Anvisa tem disponível no site o Perguntas e Respostas sobre a RDC 73/16. Este documento tem o intuito de esclarecer as empresas quanto aos questionamentos comuns da RDC 73/16. Caso alguma dúvida não esteja contemplada no material, a empresa poderá entrar em contato via Fale Conosco pelo site da Anvisa.
Art. 34. O PATE poderá ser divulgado de acordo com os critérios a serem estabelecidos pela Anvisa, resguardadas as informações sigilosas.
Este procedimento ainda não está acontecendo. Porém sempre oriento a deixar destaque no PATE quais informações a empresa entende como confidenciais.
Art. 35. As petições de pós-registro contempladas no escopo deste regulamento protocoladas antes da data vigência desta Resolução, incluindo as que se encontram em análise na Gerência-Geral de Medicamentos, serão analisadas conforme as Resoluções vigentes à época do protocolo.
§ 1º As petições já protocoladas, das quais a análise não tenha sido iniciada, cujo objeto seja enquadrado por este regulamento como de implementação imediata a serem submetidas no HMP poderão ser implementadas seguindo o disposto no art. 6º, desde que seja solicitada a desistência da petição protocolada.
§ 2º As petições já protocoladas, das quais a análise não tenha sido iniciada, cujo objeto seja enquadrado por este regulamento como de implementação imediata e que não sejam peticionadas via HMP poderão ser implementadas seguindo o disposto no art. 6°, desde que haja a formalização da mudança realizada por meio de aditamento específico ao expediente referente à mudança pós-registro, contemplando os seguintes documentos:
I - Identificação do objeto da petição e reclassificação nos termos do anexo I deste regulamento.
II - Documentação complementar requerida neste regulamento.
Este artigo auxiliou na fase de transição, pois algumas mudanças que anteriormente necessitavam aguardar avaliação prévia da Anvisa passaram a ser de implementação imediata. A regra geral é de que cada petição seria avaliada conforme a legislação vigente na época, no caso a RDC 48/09, porém houve esta abertura para transição de algumas petições para a RDC 73/16. Todos sabemos o quanto a fila de análise da Anvisa estava extensa e quanto demorava uma análise com o parecer desta Agência. Portanto, esta transição foi benéfica não somente para as empresas, mas para a Anvisa também.
Art. 36. Quando for constatada irregularidade nas petições de implementação imediata, a empresa poderá ser suspensa da realização do procedimento simplificado de mudanças pós-registro.
§ 1º Considera-se irregularidade a ausência das provas requeridas ou com prova reprovada para a mudança na data de implementação, conforme disposto no anexo I desta Resolução.
§ 2º A empresa suspensa do procedimento simplificado fica impedida por 1 (um) ano, a partir da data de publicação da decisão de suspensão, de implementar modificações pós-registro sem a autorização prévia da Anvisa, para qualquer medicamento de sua titularidade.
Quando a RDC 73 ficou vigente, em novembro de 2016, a Anvisa dava posicionamentos sobre os enquadramentos, já que muitas mudanças eram novidades na nova norma. Portanto, houve um período de transição, sem a punição de possíveis enquadramentos equivocados. A partir de 2018 entramos na fase punitiva desta forma como será comentado ao final. É muito importante a empresa estar certa do enquadramento, pois se houver irregularidades todo o portfólio da empresa ficará um ano sem ter mudanças de implementação imediata.
Art. 37. Quando a petição de renovação de registro estiver em fase recursal, não será aplicável o procedimento simplificado para as petições pós-registro do processo correspondente.
Para estes casos, a empresa protocolará o HMP com o assunto Histórico de Mudanças do Produto SEM inclusão de modificação exclusiva HMP, já que neste caso não serão permitidas mudanças de implementação imediata.
Art. 38. Fica revogada a Resolução da Diretoria Colegiada - RDC nº. 48, de 06 de outubro 2009 e a Instrução Normativa nº. 11, de 06 de outubro de 2009.
Art. 39. Os itens 3.1.2, 3.1.3, 3.2 e 3.4 do Anexo da Instrução Normativa nº. 2, de 30 de março de 2009, publicada no Diário Oficial da União de 01/04/2009, passam a vigorar com a seguinte redação:
"3.1.
3.1.2. No caso de sólidos, deverá ser considerada a quantidade mínima de 100.000 unidades farmacotécnicas ou 10% do lote industrial, a que for maior. (NR)
3.1.3. Lotes de sólidos menores que 100.000 unidades farmacotécnicas poderão ser apresentados para fins de registro e pós-registro desde que seu tamanho corresponda ao do lote industrial pretendido. (NR)
3.2. Para mudanças de tamanho de lote, a empresa deverá seguir a norma específica de alterações pós-registro." (NR)
3.4. Para produtos cuja concentração do princípio ativo em relação à fórmula seja inferior a 2% (dois por cento), não serão permitidos lotes pilotos com quantitativos diferentes dos lotes industriais.
A alteração da IN2/09 também foi um marco neste momento, pois esta instrução normativa considerava a quantidade mínima de 50 mil unidades farmacotécnicas como tamanho mínimo de um lote piloto. Aqui foi alterada para o mínimo de 100 mil unidades farmacotécnicas.
Art. 40. Serão aceitos lotes pilotos para fins de registro e pós-registro de sólidos entre 50.000 e 100.000 unidades farmacotécnicas, desde que fabricados anteriormente à vigência dessa resolução e cuja petição seja protocolada até 01 (um) ano a partir da vigência dessa Resolução.
Parágrafo único. Lotes pilotos cuja concentração do princípio ativo seja inferior a 2% (dois por cento) e superior a 0,99 miligramas por unidade posológica em relação a fórmula serão aceitos para fins de registro e pós-registro de sólidos desde que fabricados anteriormente à vigência dessa resolução e cuja petição seja protocolada até 01 (um) ano a partir da vigência dessa.
Este artigo representa a fase de transição da definição da quantidade mínima para lote piloto sendo 100 mil unidades farmacotécnicas.
Art. 41. Para produtos registrados com lotes pilotos de sólidos fabricados entre 50.000 e 100.000 unidades farmacotécnicas será permitida a implementação imediata do aumento do tamanho de lote em até 10 (dez) vezes, mediante protocolo individual com código de assunto específico, atendendo as seguintes condições:
I - O peticionamento, para produtos registrados antes da vigência dessa norma, deverá ocorrer no prazo máximo de 2 (dois) anos da vigência da norma;
II - O peticionamento, para produtos registrados após a vigência dessa norma, deverá ocorrer no prazo máximo de 2 (dois) anos contados a partir da concessão do registro, não podendo exceder 5 (cinco) anos da vigência da norma;
III - A petição deverá conter o cronograma do estudo de biodisponibilidade relativa/bioequivalência e os documentos previstos na modificação f, do item 6 (seis), anexo I.
O relatório técnico de biodisponibilidade relativa/bioequivalência deverá ser apresentado no prazo máximo de 2 (dois) anos após o peticionamento.
Parágrafo único: A não apresentação do estudo de biodisponibilidade relativa/bioequivalência nos termos art. 41 acarretará no cancelamento do registro.
A Anvisa disponibilizou um quadro resumindo como será esta inclusão de tamanho de lote no Perguntas e Respostas da RDC 73/16:
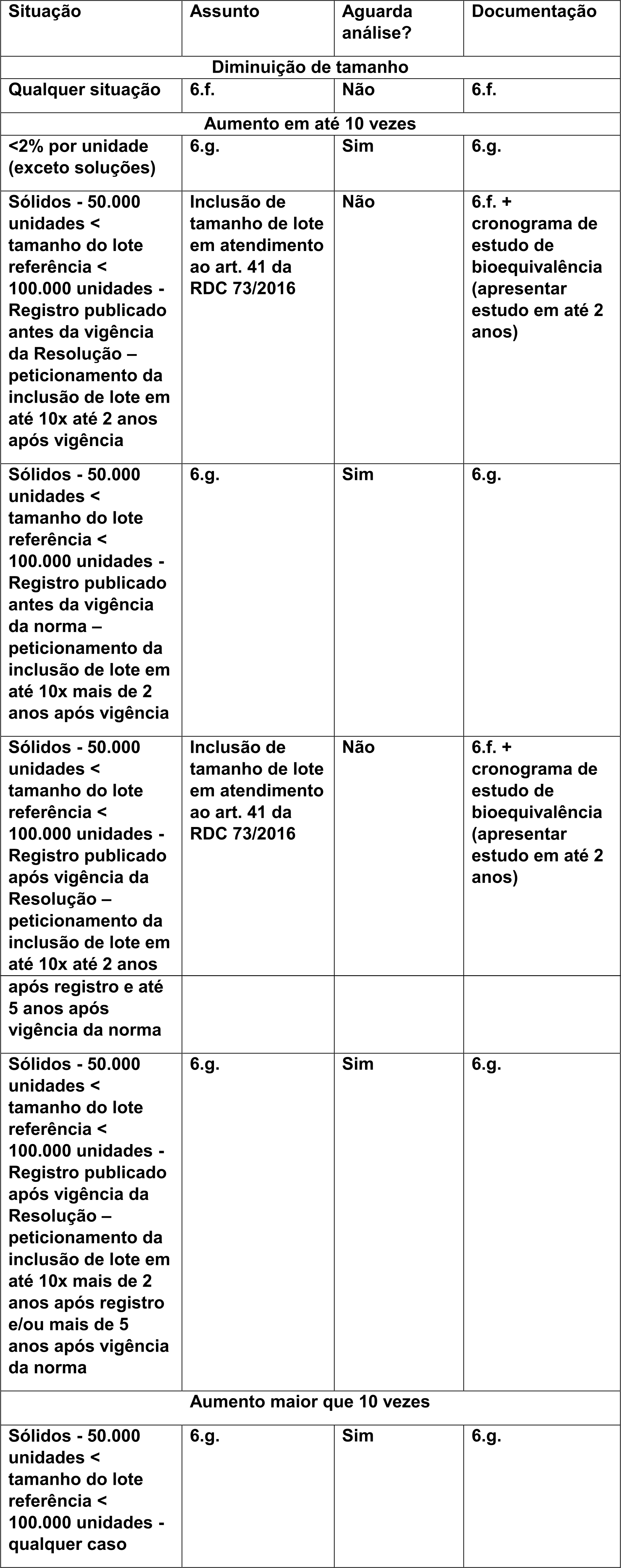
Neste mesmo documento de Perguntas e Respostas, a Anvisa apresenta um caso hipotético para melhor exemplificar os casos que precisam ou não de estudo de bioequivalência:
“Produto registrado em 2009 com tamanho de 70.000 unidades. Em 2011, houve inclusão de tamanho de lote para 600.000 unidades, com todas as provas de acordo com a RDC 48/09. Não há nenhuma outra mudança pós-registro. Com a vigência da RDC 73/16 por si só, nenhuma ação será requerida. Entretanto, se a empresa pretende aumentar o tamanho de lote para 700.000 unidades na vigência da RDC 73/16, o lote de referência é de 70.000. Para este tipo de mudança, a empresa deverá peticionar conforme item 6.g. Alternativamente, a mudança poderá ser enquadrada no art. 41 se seus requisitos forem cumpridos.”
Art. 42. Os artigos 19, 20 e 21 da Resolução da Diretoria Colegiada - RDC nº. 47, de 08 de setembro de 2009, publicada no Diário Oficial da União de 09/09/2009, republicada em 19 de janeiro de 2010, passam a vigorar com a seguinte redação:
"Art. 19. As alterações das informações dispostas em bula dos medicamentos que não possuem Bula Padrão decorrentes de uma mudança pós-registro devem ser disponibilizadas concomitantemente à implementação da mudança.
Parágrafo único. As novas versões de bulas deverão ser submetidas por meio de notificação de alteração de texto de bula via peticionamento eletrônico em até 30 dias da aprovação, contendo as informações das últimas bulas publicadas no Bulário acrescidas das informações aprovadas nesta petição." (NR)
"Art. 20. Para as alterações nos textos de bulas dos medicamentos que possuem Bula Padrão, vinculadas às alterações de suas respectivas Bulas Padrão, exceto para as informações específicas do produto, as bulas devem ser notificadas eletronicamente em até 90 (noventa) dias e disponibilizadas em até 180 (cento e oitenta) dias após a publicação das Bulas Padrão no Bulário Eletrônico, devendo ser implementadas, independentemente de manifestação prévia da Anvisa.
Parágrafo único. As empresas devem avaliar se as mudanças relacionadas à posologia, ampliação de uso, inclusão de nova via de administração e/ou nova indicação terapêutica são aplicáveis ao seu produto. Caso não sejam, não há a obrigatoriedade de cumprimento do prazo do caput e o prazo será avaliado caso a caso pela Anvisa, dependendo da(s) alteração(ões) pós-registro que será(ão) necessária(s) para a adequação do produto." (NR)
"Art. 21. As alterações das informações dispostas em bula dos medicamentos genéricos e similares decorrentes de uma mudança pós-registro devem ser disponibilizadas concomitantemente à implementação da mudança.
Parágrafo único. As novas versões de bulas deverão ser submetidas por meio de notificação de alteração de texto de bula via peticionamento eletrônico em até 30 dias da aprovação, contendo as informações" (NR)
Neste artigo referente às bulas, informo que também devem ser observadas além da RDC 47/09, a RDC 60/12 que dispõe sobre os procedimentos no âmbito da Anvisa para alterações de textos de bulas de medicamentos e dá outras providências e o Guia de Submissão Eletrônica de Texto de Bula.
Art. 43. O artigo 76 da Resolução da Diretoria colegiada RDC nº. 71, de 22 de dezembro de 2009, publicada no Diário Oficial da União de 23/12/2009, passa a vigorar com a seguinte redação:
"Art. 76. As alterações das informações dispostas na rotulagem decorrentes de uma mudança pós-registro devem ser disponibilizadas concomitantemente à implementação da mudança.
Parágrafo único. Os novos modelos de rotulagem deverão ser submetidos por meio de notificação de rotulagem via peticionamento eletrônico em até 30 dias da aprovação, contendo o modelo mais recente de rotulagem já peticionado e a alteração das informações aprovadas nesta petição." (NR)
Neste artigo referente às rotulagens, informo que também devem ser observadas além da RDC 71/09, a RDC 61/12 que dispõe sobre os procedimentos no âmbito da Anvisa para alterações de rotulagens de medicamentos e dá outras providências.
Art. 44. O descumprimento das disposições contidas nesta Resolução constitui infração sanitária, nos termos da Lei nº. 6.437, de 20 de agosto de 1977, sem prejuízo das responsabilidades civil, administrativa e penal cabíveis.
Art. 45. Os efeitos do § 2º do art. 36 passarão a vigorar no prazo de 360 (trezentos e sessenta dias) dias, contados a partir da vigência desta Resolução.
A vigência da RDC 73 ocorreu em novembro de 2016, portanto, a fase punitiva da legislação iniciou-se um ano após, em novembro de 2017.
Art. 46. Esta Resolução entra em vigor em 120 (cento e vinte) dias a partir da data de sua publicação.
Esta norma foi publicada no DOU de 8 de abril de 2016, portanto, considerando este artigo, a vigência seria em agosto de 2016. Porém este prazo foi prorrogado por mais 90 dias conforme RDC 100/16 e, portanto, a vigência da RDC 73/16 ocorreu em novembro de 2016.
JARBAS BARBOSA DA SILVA JR.
Diretor-Presidente
ANEXO I
1. MUDANÇAS RELACIONADAS AO INSUMO FARMACÊUTICO ATIVO:
Desmembrada em cinco itens, a principal novidade é a mudança B. Substituição ou inclusão de local de fabricação do IFA do mesmo grupo farmoquímico, que é de implementação imediata após protocolo e o item D. Mudança menor de produção do IFA, que também é de implementação imediata, porém com anotação em HMP. Para este último caso, estão inclusas as mudanças de tamanho de lote do IFA que antigamente não era prevista pela RDC 48/09.
Além disso, um dos pontos mais importantes das mudanças relacionadas ao IFA são as mudanças C. Substituição ou inclusão de novo fabricante do IFA e E. Mudança maior de produção do IFA que uma das solicitações é a apresentação do Relatório técnico de estudo de biodisponibilidade relativa/bioequivalência do medicamento. Apesar de tudo, esta prova pode ser substituída se não houver alterações em propriedades físico-químicas do IFA com potencial impacto em biodisponibilidade.
2. MUDANÇAS RELACIONADAS AOS TESTES, LIMITES DE ESPECIFICAÇÕES E MÉTODOS ANALÍTICOS DO CONTROLE DE QUALIDADE E ESTABILIDADE DO INSUMO FARMACÊUTICO
ATIVO E MEDICAMENTO
Além de trazer oito itens de mudança, a novidade aqui é a aplicabilidade das mudanças para o insumo farmacêutico ativo. Conforme dito anteriormente, a RDC 48/09 regulamentava como mudança pós-registro apenas mudanças de métodos analíticos para produto acabado.
3. MUDANÇAS RELACIONADAS AOS TESTES, LIMITES DE ESPECIFICAÇÕES E MÉTODOS DO CONTROLE DE QUALIDADE DO EXCIPIENTE
Este tipo de mudança também é novidade e, apesar de ter apenas um item para enquadramento e ser de implementação imediata com anotação em HMP, causou um impacto grande, pois geralmente as empresas têm muitos excipientes que são utilizados nos medicamentos e, apesar das empresas terem o controle das atualizações de métodos destas matérias-primas, não era considerada uma mudança pós-registro.
4. MUDANÇAS DE DESCRIÇÃO E COMPOSIÇÃO DO MEDICAMENTO
Com 14 possíveis enquadramentos, este tipo de mudança está bem desmembrada também. O interessante é a mudança A. alteração de formato e dimensões de comprimidos, cápsulas, supositórios e óvulos. Para a alteração do formato faz-se necessária a alteração de punções. Este tipo de mudança não ficava clara na RDC 48/09 e como não vinha de atualização farmacopeica nem era estreitamento de faixa (únicas alternativas para um enquadramento de implementação imediata), era necessário protocolar e aguardar deferimento. Agora, com a RDC 73/16 a implementação é imediata se não houver alteração qualitativa e quantitativa da composição, do peso médio, das demais especificações e das características de desempenho do produto.
Importante ressaltar nas mudanças de composição do medicamento, o fato de que não há mais mudanças moderadas de excipientes, porém, o quadro que traz os limites de alteração de cada tipo de excipiente para os enquadramentos traz a alteração moderada de excipiente. Vamos lá, não há assunto de enquadramento para mudanças moderadas de excipientes, porém se o enquadramento for este, a mudança fica isenta da apresentação de estudo de bioequivalência.
5. MUDANÇAS RELACIONADAS AO LOCAL DE UMA OU MAIS ETAPAS DO PROCESSO PRODUTIVO DO MEDICAMENTO
Com oito itens para enquadramentos, este tipo de mudança trouxe um ganho muito grande para o setor. Imagine que a empresa tenha que fazer uma alteração de local de fabricação do medicamento, seja por expansão da empresa ou por relações comerciais... este tipo de mudança aguardava deferimento para ser implementada. Atualmente, com a RDC 73/16, para medicamentos de liberação convencional esta mudança é de implementação imediata após o protocolo. Para medicamento de liberação modificada, há o enquadramento como mudança menor se não houver alteração de endereço, esta mudança menor também é de implementação imediata após protocolo na Anvisa.
Um dos pontos mais importantes aqui é o fato da Anvisa ter classificado que para medicamentos estéreis não há mudanças menores. Para esta categoria de medicamentos, a mudança de local de fabricação em partes ou em sua totalidade deve aguardar deferimento da Anvisa para implementação.
Referente a documentos a serem apresentados, a principal diferença está na apresentação de relatório técnico de estudo de biodisponibilidade relativa/bioequivalência para mudanças classificadas como maiores.
Uma novidade neste tipo de mudança foi também o item H. Inclusão ou substituição de local de controle de qualidade, apesar de ter entrado agora na norma, já se esperava algo do tipo, pois algumas discussões sobre terceirizações já apontavam para o entendimento de que em alguns casos deveria ser tratado como pós-registro.
6. MUDANÇAS RELACIONADAS AO PROCESSO DE PRODUÇÃO DO MEDICAMENTO, EQUIPAMENTO E TAMANHO DE LOTE
O que mais chama atenção neste tipo de mudança são as formas de enquadramentos, por exemplo, a RDC 48/09 trazia como mudança menor de processo, as mudanças de velocidade, temperatura, tempo e ordem de adição dos componentes da fórmula. Atualmente pela RDC 73/16 não importa se as mudanças foram somente estas, o que importa é se elas foram realizadas em parâmetros ou etapas críticas ou não críticas do processo produtivo, sendo que, a validação de processo que define quais parâmetros e etapas é considerada crítica.
Um ponto interessante de se analisar é que, pela RDC 73/16, há a possibilidade de mudanças de equipamentos com diferente desenho e princípio de funcionamento, sendo de implementação imediata deste que a mudança ocorra com um equipamento que não participe de etapas críticas do processo produtivo. Este tipo de mudança pela RDC 48/09 aguardava deferimento para implementar, então, passavam-se dois, as vezes, três anos aguardando.
Com relação aos documentos que devem ser apresentados para mudanças maiores de processo produtivo e mudanças maiores de tamanho de lote, deve-se apresentar relatório técnico de estudo de biodisponibilidade relativa/bioequivalência ou justificar a ausência do estudo.
7. MUDANÇAS RELACIONADAS À EMBALAGEM DO MEDICAMENTO
Este tipo de mudança, na RDC 48/09, era tratada apenas como inclusão de novo acondicionamento apenas. A RDC 73/16 desmembrou este tipo de mudança em 12 possíveis enquadramentos, prevendo ainda, a mudança relacionada ao controle de qualidade da embalagem.
Um ponto muito importante aqui é a solicitação de estudos referente à comprovação de que não ocorre interação entre a embalagem e seu conteúdo como migração dos componentes do material proposto para o conteúdo e perda dos componentes do medicamento na embalagem, o que chamamos de extraíveis e lixiviáveis.
8. INCLUSÃO DE NOVA APRESENTAÇÃO
A principal observação para esta mudança é também a mais óbvia: “A nova apresentação deverá ser condizente com a posologia do medicamento e duração do tratamento.” Com a dinâmica de uso racional de medicamentos, a empresa deve projetar suas apresentações de forma condizente à posologia do produto.
9. MUDANÇAS RELACIONADAS AO PRAZO DE VALIDADE OU AOS CUIDADOS DE CONSERVAÇÃO DO MEDICAMENTO
Nesta mudança, chamo a atenção para a mudança de redução do prazo de validade. Esta mudança está prevista como sendo de implementação imediata, porém após protocolo. Deve ser apresentado estudo de estabilidade demonstrando que o produto não é estável no prazo de validade registrado. Nesta situação a empresa deve realizar o protocolo imediatamente após a identificação desta instabilidade. Por isso o registro de um produto pode iniciar com prazo de validade de 24 meses, por exemplo, afinal o estudo de estabilidade acelerado completo e longa duração parcial (6 meses) dá subsídios para o registro com este prazo de validade. Porém, durante a condução dos estudos, este prazo pode reduzir para 18 meses, por exemplo, no caso de instabilidade.
10. INCLUSÃO DE NOVA CONCENTRAÇÃO
Estas mudanças seguem os dispositivos da RDC 200/17 de Registro de Medicamentos. Por mais que esteja classificada como pós-registro, a avaliação é da Gerência de Segurança e Eficácia da Anvisa.
11. MUDANÇAS RELACIONADAS À POSOLOGIA, AMPLIAÇÃO DE USO, INCLUSÃO DE NOVA VIA DE ADMINISTRAÇÃO, NOVA INDICAÇÃO TERAPÊUTICA
Aqui coloco a mesma observação que apontada no item anterior, por mais que esteja classificada como pós-registro, a avaliação é da Gerência de Segurança e Eficácia da Anvisa.
12. MUDANÇAS RELACIONADAS AO NOME DO MEDICAMENTO, CANCELAMENTO DO REGISTRO DO MEDICAMENTO e EXCLUSÃO DE LOCAL DE FABRICAÇÃO DO FÁRMACO, LOCAL DE EMBALAGEM PRIMÁRIA, LOCAL DE EMBALAGEM SECUNDÁRIA E/OU LOCAL DE FABRICAÇÃO DO PRODUTO
Com relação ao nome do medicamento, é preciso apresentar uma Declaração de não comercialização do produto. Uma vez o produto comercializado, não há possibilidade de solicitação de troca de nome (marca).
