





A atualização da legislação sanitária nacional (RDC 17/10) tornou-se um desejo de longa data. Para aqueles que participam de inspeções para exportação era perceptível a dificuldade de aceitação da regulamentação nacional frente a outras mundialmente utilizadas. Com o objetivo de harmonizar os padrões utilizados nas inspeções sanitárias praticadas em outros países, a Agência Nacional de Vigilância Sanitária (Anvisa) buscou tornar o Brasil membro do Pharmaceutical Inspection Co-operation Scheme (PIC/s).
Para que a adesão fosse possível, a Anvisa se propôs a atualizar nossa regulamentação sanitária, que trata de Boas Práticas de Fabricação para Medicamentos. Dessa maneira, surgiu a nova RDC 301/19 e seus anexos.
RDC 301/19 - Cenário prático para a seleção de pior caso e limites físico químicos, avaliados sob o risco toxicológico requerido na nova regulamentação sanitária brasileira.
Divulgada em agosto de 2019 e vigente desde outubro do mesmo ano, a regulamentação trata fortemente do cuidado com os riscos relacionados á contaminação cruzada, seja na gestão dos riscos dos processos, no compartilhamento de área ou mesmo na validação de limpeza. O assunto tomou mais espaço no texto de redação da nova normativa e maior dimensão nas discussões na comunidade farmacêutica.
Dentre os assuntos relacionados que envolvem a contaminação cruzada, um dos mais impactados foi a validação de limpeza.
O estudo busca constatar a qualidade do procedimento de limpeza, sob o seguinte aspecto: se o procedimento de limpeza da rota consegue limpar, com qualidade, o produto chamado de pior caso, todos os outros serão seguramente limpos.
O estudo de validação de limpeza envolve a avaliação da eficácia da limpeza padronizada e praticada ao término de cada lote fabricado, ou mesmo ao término de uma campanha, a fim de avaliar se possíveis residuais físico-químicos (ativo ou agente de limpeza) e microbiológicos estão em uma concentração aceitável. Isso quer dizer que, caso exista algum residual presente na rota, ele deve se manter em uma concentração segura (dentro do critério de aceitação para a linha). Isso explicaria que o residual, mesmo este seja carreado ao produto subsequente, ainda assim, não demonstraria risco ao paciente.
A validação de limpeza pode contemplar duas modalidades de estudo: Lote a Lote e Campanha.
Estudo Lote a Lote: ocorre quando o momento de limpeza de uma rota ou equipamento é praticado entre a fabricação de cada lote produzido, ou seja, ao final de cada lote, há uma limpeza completa.
Estudo em Campanha: ocorre na fabricação consecutiva dos lotes de um mesmo produto ou similares entre si, e para essa modalidade de estudo não se aplica o conceito de seleção de pior caso, sendo realizado para todos os produtos que a empresa necessitar de produção em campanha. Deve-se mencionar tempo e número de lotes a ser estudado.
Nas áreas de fabricação, as linhas de produção onde são compartilhados os mesmos equipamentos e áreas por uma quantidade aleatória de diferentes produtos é denominada linha ou rota multipropósito.
Alguns pontos importantes devem ser observados como premissas para o início de estudo de validação de limpeza, são eles:
- Desenvolver um procedimento de limpeza apropriado aos equipamentos, utensílios e mangueiras que tenham contato direto com produto. Esse procedimento é determinado com base nas características do equipamento e linha, características produto/ formulação/ativo, características e risco do processo e técnica de limpeza utilizada - manual, Clean Open Place (CIP), Clean Open/Out Place (COP) etc.;
- Elaborar e tornar vigente o procedimento operacional padrão (POP) de limpeza e operação dos equipamentos e linha, conforme padrão do Sistema de Qualidade Farmacêutica (SQF) da empresa;
- Ter concluídos e vigentes os documentos técnicos do SQF, entre eles: Manual e política de Qualidade, POP de elaboração, guarda e descontinuidade de documentos controlados, POP para Controle de Mudanças, abertura e tratamento de Desvios, Plano Mestre de Validação (PMV), POP e Plano de calibração, manutenção corretiva e preventiva, POP para elaboração de documentos e execução das qualificações, amostragem e execução da validação de limpeza, POP de validação de Métodos Analíticos, POP para Qualificação e Validação em Sistema de Utilidades, POP para Validação de Sistemas Computadorizados etc.;
- Avaliar e definir se a linha é dedicada ou multipropósito;
- Avaliar e definir se a limpeza completa é feita ao término de cada lote ou se é um estudo em campanha, onde temos uma limpeza parcial entre os lotes e uma limpeza completa ao término do último lote do produto em campanha;
- Instrumentos calibrados;
- Equipamentos e instalações qualificados;
- Validação dos sistemas computadorizados envolvidos na operação e limpeza da linha de fabricação, bem como das utilidades e do suporte analítico envolvido no estudo;
- Validação das utilidades críticas que participam da fabricação e análise abrangidas no estudo (ar comprimido, nitrogênio, ar mandado, HVAC, água purificada e água para injetáveis);
- Realizar a seleção do pior caso para linha e rota;
- Fazer a seleção dos tipos de amostragens (swab ou rinsagem), conforme avaliação de risco da amostragem envolvida no processo e produto. É preciso recomendar, nessa etapa, o tipo de swab, volume e tipo de solvente a ser empregado na rinsagem;
- Desenvolver a seleção dos pontos de amostragem conforme avaliação de risco relacionado ao processo e produto;
- Levantar a área ou superfície dos equipamentos e linha que possuem contato direto com o produto;
- Desenvolver e validar os métodos analíticos a serem utilizados para o residual microbiológico e físico químico (ativo, agente de limpeza e sanitizante, caso haja), incluindo, também, a validação da recuperação para as amostragens dos resíduos avaliados;
- Pessoal envolvido nas atividades em geral deve ser treinado e qualificado;
- Solicitar a avaliação dos dados toxicológicos das substâncias envolvidas no estudo por meio do valor PDE;
- Elaborar e aprovar o protocolo de validação de limpeza, constando plano de amostragem, linha, produtos da linha, produto pior caso ou campanha a ser estudada, equipamentos, utensílios e mangueiras utilizados no processo de fabricação (que estão relacionados ao estudo a ser realizado), pontos de amostragem, tipo de amostragem, cálculos utilizados na obtenção dos limites dos residuais, limites para cada amostra a ser coletada e outras observações pertinentes ao estudo;
- Planejar, junto à área de programação em produção, o momento de fabricação de, no mínimo, três lotes consecutivos do produto pior caso, para estudos Lote a Lote, ou mínimo de três campanhas do produto em estudo;
- Lembrando que sempre antes das amostragens físico-químicas ou microbiológicas devem ser realizadas as inspeções visuais, ou seja, aplicar o critério de visualmente limpo. Caso seja observado algum resíduo, reprovar a amostragem e rever o processo de limpeza, antes de iniciar uma nova amostragem.
- Proceder a amostragem e o envio ao laboratório para subsequente análise, ao término do processo de fabricação do produto pior caso, ou mesmo entre e após os lotes de uma campanha em estudo. Caso não exista um estudo justificado sobre o tempo de guarda das amostras, é indicado que elas sejam analisadas imediatamente após o envio ao laboratório;
- Estudos complementares ainda devem ser avaliados, como por exemplo, tempo de equipamento sujo e tempo de equipamento limpo; e
- Após todos os resultados das amostragens estarem concluídos e aprovados, um relatório final deve ser elaborado e aprovado. Dessa maneira, conclui-se um estudo de validação de limpeza.
Seleção do pior caso (worst case).
Dentro da avaliação de risco aplicada à validação de limpeza, alguns pontos geraram grande discussão no meio farmacêutico, principalmente porque causam impacto na estratégia de validação de limpeza que as empresas já utilizam, e demoraram a conquistar. Isso pode gerar custos altos nas adequações e revalidações, que são justificados pela alteração solicitada na regulamentação. No entanto, todo esse cenário de adequação pode ficar pior, caso venha, agregado a ele, a falta de conhecimento técnico e falta de planejamento.
Para o início de uma avaliação estratégica é necessário verificar a existência de um levantamento lógico e robusto dos produtos que fazem parte da linha ou rota em estudo. Recomenda-se uma planilha que facilite a gestão das informações, envolvendo tratamento matemático para o pior caso, e ainda, se necessário, um desempate.
Nessa etapa seriam envolvidas informações importantes, como por exemplo: nome do produto, posologia de administração, tamanho de lote comercial, máxima dose diária, dificuldade de limpeza, toxicidade, solubilidade do ativo em água e poderia ainda ser incluída a rota de equipamentos e utensílios, descrevendo a área de contato de cada um deles. Esse perfil seria o mais recomendado pela Anvisa.
Porém, ainda podemos encontrar outro modelo de racional para a seleção do pior caso, aplicando o Fator de Ocupação, em que as empresas se baseiam no cálculo do Worst Case Index (WCI) - muito utilizado nas inspeções internacionais. Nesse modelo são utilizados, em conjunto, o Fator de Ocupação, o Fator de Solubilidade e o Fator de Dificuldade de Remoção, exemplificados nas tabelas I, II e III.
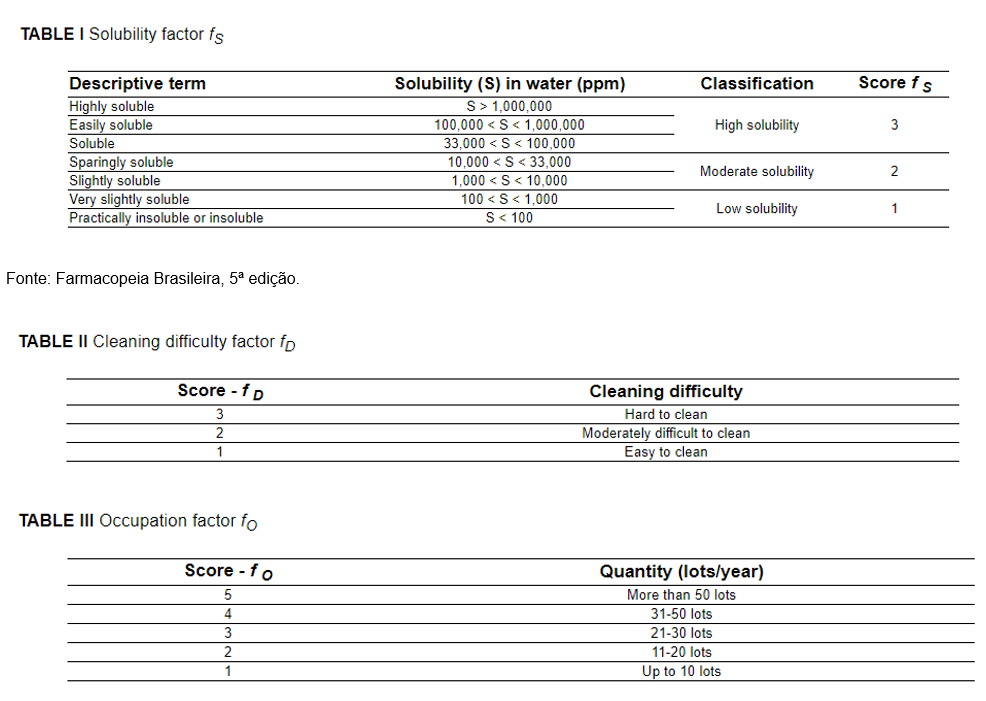
Após a avaliação desses, aplica-se o cálculo descrito abaixo:
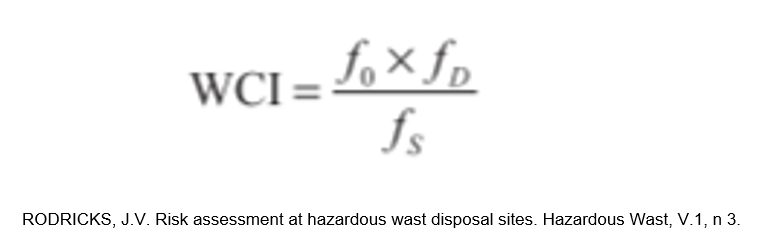
Sobre a alteração descrita na nova regulamentação, a aplicação técnica da avaliação toxicológica, antes utilizada por meio de valores descritos por fichas de segurança para substâncias químicas (FISPQs) ou de Dose Letal (DL50%), geravam muitas dúvidas em relação à interpretação, como por exemplo, sobre as diferentes matrizes (tipos de animais) utilizados nos estudos em que seriam comparados os valores finais dos produtos e a disponibilidade dos valores descritos em FISPQs, pois nem todas as substâncias químicas tinham tal informação disponível.
A RDC 301/19 trouxe uma grande mudança nessa etapa de validação, sendo necessária, atualmente, a inclusão da avaliação do permitted daily exposure (PDE) para os fármacos produzidos em cada rota produtiva validada. Essa informação visa demonstrar uma maior preocupação com o consumidor final do produto, uma vez que a avaliação toxicológica para os produtos será item crucial aplicado a novos limites dos estudos de validação de limpeza.
A avaliação de PDE envolve a análise e aplicação dos conceitos de toxicologia para o melhor entendimento do processo. Os princípios para cálculo do PDE podem ser encontrados no EMA’s “Guideline on setting health based exposure limits for use in risk identification in the manufacture of diferent medicinal produts in shared facilities (EMA, 2014; PIC/S, 2018 a.b) e no Annex 15 Qualification and Validation (PIC/S, 2018).
Novo comparativo
Nesse novo cenário, a melhor estratégia aplicada aos estudos já existentes é realizar um novo comparativo entre todos os produtos da linha, substituindo o valor utilizado anteriormente (LD50% ou valor obtido da classificação toxicológica da FISPQ) pelo valor atual obtido na avaliação toxicológica PDE. A partir dessa substituição, deve-se recalcular os valores finais de pontuação para cada produto, em que o produto com maior pontuação seria o selecionado então como o pior caso.
Se o produto pior caso não permanecer o mesmo, a estratégia de adequação deve partir para a revalidação completa, iniciando da validação da metodologia analítica relacionada aos residuais das substâncias físico-químicas. O item seguinte deste artigo trata do modelo que deve ser praticado aos novos estudos.
Sob o ponto de vista toxicológico, existe a recomendação da avaliação do pior caso realizada num comparativo entre os diferentes cenários obtidos com a conjugação dos produtos: um possível antecessor e um possível sucessor, tratando de situações hipotéticas de fabricação. Isso será detalhado no próximo item deste artigo.
A partir da seleção do pior caso, partimos para a etapa documental, na elaboração do protocolo, com a análise de risco sendo aplicada, então, a seleção dos tipos de amostragem e pontos a serem amostrados. A escolha da melhor ferramenta a ser utilizada é da empresa. Não existe, nesse momento, uma recomendação de uso de um modelo único.
A adequação dos estudos de validação de limpeza tem prazo de transitoriedade regulamentado, segue descrito abaixo:
- 6 meses – Reestruturação SQF
- 12 meses – Inclusão de novos produtos
- 24 meses – 30% do portifólio de produtos
- 36 meses – 60% do portifólio de produtos
- 48 meses - 100% do portifólio de produtos
Aplicação dos cálculos toxicológicos nos novos estudos de validação de limpeza
Os cálculos aplicados, atualmente, para resíduos de agente de limpeza ainda são os mesmos recomendados pelo Guia de Assuntos Relacionados à Garantia de Qualidade (Anvisa/2006). Esses cálculos seguem descritos abaixo:
- Critério de 10 ppm, em que a fórmula utilizada é:
MAR (mg/cm2) = (R x MBSt)
SSA
- Critério de toxicidade, em que a fórmula utilizada é:
ADI (mg) = (LD50 x 70 ou 12Kg)
(2000 x SF)
MACO (mg/cm2) = (ADI x MBSt)
(LDDf x SSA)
Dados:
- MBSt = mínimo tamanho de lote do produto subsequente, em que:
Critério 10 ppm - unidade em mg (sólidos e líquidos).
Critério de Toxicidade - unidade em mg para sólidos e mL para líquidos.
- R = 10 mg/1.000.000 mg.
- SSA = área total da superfície de contato do equipamento com o produto na nota de produção (cm2).
- 70 Kg ou 12 Kg = 70 kg é o peso praticado para produto de uso adulto; 12 Kg é o peso praticado para produto de uso pediátrico.
- SF = fator de segurança 1000.
- ADI = ingestão diária aceitável.
- MACO = máximo resíduo permitido.
Para os cálculos relacionados a resíduos de ativos em estudos já existentes, é recomendado que seja realizado um comparativo entre os cálculos existentes, conforme o Guia da Anvisa e o cálculo aplicando o valor de PDE, e verificar, então, se os resultados foram superiores ou inferiores aos limites obtidos inicialmente.
Caso os limites encontrados, aplicando o valor de PDE, forem superiores aos obtidos inicialmente, deve-se registrar a avaliação realizada e justificar que não ocorreu impacto direto no estudo validado.
Caso os limites encontrados no cálculo, aplicando o valor de PDE, forem inferiores aos obtidos inicialmente, deve-se fazer um levantamento dos resultados analíticos já obtidos e compará-los com o novo critério de aprovação. Os resultados fora da nova especificação reprovam o teste, quando é requerida a revalidação completa.
- Ponto de vista toxicológico
A regulamentação atual traz alguns pontos importantes do ponto de vista toxicológico, que devem ser acrescentados na avaliação dos novos estudos para a seleção do pior caso - worst case - da linha produtiva, sendo eles:
- Qual o produto anterior;
- Qual a via de administração do produto anterior;
- Qual a via de administração do produto subsequente;
- Avaliação dos dados clínicos de cada produto;
- Avaliação dos dados e estudos não clínicos de cada produto.
Segundo o farmacêutico especialista em toxicologia, Leandro Scorsi, também devemos considerar alguns fatores relacionados à fabricação dos fármacos em equipamentos e rotas compartilhados, sendo eles:
- Se existem relatos na literatura técnica sobre o assunto, que menciona o risco sobre a substância gerada a partir da interação entre a droga anterior e a posterior;
- Se essa interação citada acima pode gerar algum composto tóxico;
- Qual o mecanismo de distribuição do agente tóxico pelo organismo;
- Qual a característica físico-química que esse agente tóxico possui (lipossolúvel ou hidrossolúvel).
Encontramos, em algumas publicações, a discussão do cálculo de PDE de um produto baseado na escolha da via de administração do fármaco, tendo seus valores identificados em cada caso.
Atualmente, na indústria farmacêutica, não é comum a avaliação da combinação de fabricação de uma campanha produtiva. Assim, a partir desse momento é um aspecto importante para a discussão do racional do estudo vigente ou proposto.
Outro ponto que devemos ter bastante atenção, e que causará muito impacto no estudo de validação de limpeza, é a via de administração dos produtos. Muitas vezes, fabricamos na mesma rota produtiva, por exemplo, produtos de via de administração subcutânea, intramuscular e intravenosa. No caso de produto subsequente com via de administração mais crítica, o valor de PDE permitido será menor se comparado com outra via de administração com menor criticidade.
A escolha do worst case para os novos estudos ou avaliação do estudo vigente será muito mais complexa devido ao aumento do número de variáveis que devem ser avaliadas. Além disso, deve existir uma análise de risco embasando todas as escolhas, mitigações e novo design do estudo.
O cálculo do PDE envolve algumas etapas:
- Avaliação do perigo (revisão bibliográfica contendo dados de estudos clínicos e não clínicos sobre o produto);
- Identificação dos efeitos críticos para a saúde (limite mais sensível que pode gerar efeitos adversos em uma determinada faixa populacional);
- Seleção do ponto de partida (PoD – dose relacionada a efeitos críticos);
- Aplicação do fator de ajuste composto (CAF). CAF é o resultado de multiplicação de diversos fatores de ajustes de um produto.
Após a identificação dos itens acima, aplicamos os valores na seguinte equação:

Em algumas literaturas, podemos observar uma modificação na fórmula para a obtenção do valor de PDE, evidenciada da seguinte forma:
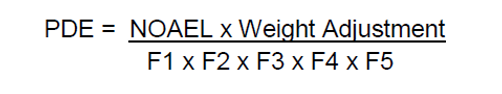
Para a equação acima, precisamos avaliar o conjunto de fatores individualmente. A seguir iremos descrever o significado de cada fator F1, F2, F3, F4 e F5.
F1: fator relacionado para extrapolação entre espécies: valor entre 2 e 12;
F2: Fator relacionado para variabilidade entre indivíduos: valor = 10;
F3: fator relacionado para estudo de toxicidade curta com repetidas doses para a droga avaliada (estudo com tempo menor do que 4 semanas): valor = 10;
F4: fator relacionado a casos de toxicidade grave da droga, ex: neurotoxicidade, carcinogenicidade, teratogenicidade: valor entre 1 e 10;
F5: fator relacionado a ausência de dados sobre o NOAEL. Quando só é encontrado o LOAEL da droga, deve ser utilizado o F5 para ajuste no cálculo: valor entre 1 e 10.
Essa avaliação de cada fator deve ser considerada por meio da análise de um toxicologista, com base na revisão bibliográfica proposta para a droga relacionada.
É importante entender que, quanto maior as notas para cada fator, menor será o limite (µg/dia) permitido para a droga em questão.
E, nesse caso, maior será a dificuldade de quantificação pelo método analítico vigente. Também será exigida uma maior qualidade e robustez no processo de limpeza dos equipamentos.
O PIC/s possui um anexo no seu guideline para uma avaliação inicial da droga a ser estudada. Esse anexo ajuda a entender, inicialmente, quais as características da droga. Segue abaixo o anexo:

Baseado na discussão e explanação acima, podemos entender o impacto que isto trará na validação de limpeza vigente.
Outro ponto importante a ser considerado é o aumento do espectro de avaliação para os produtos. A empresa deve iniciar a avaliação pelos casos considerados mais críticos ou mais complexos, como o uso de peças dedicadas para determinados produtos, tecnologia single use entre outras estratégias na validação de limpeza, que possam impactar no cálculo de PDE de cada produto.
* FERNANDA BIDO é farmacêutica, mestre em Processos Industriais pelo IPT/USP e professora do ICTQ – Instituto de Pesquisa e Pós-Graduação para o Mercado Farmacêutico e gerente de Consultoria na Azbil Telstar Brasil.



