





A equivalência farmacêutica entre dois medicamentos garante, por meio de testes in vitro, que ambos contêm o mesmo fármaco, na mesma dosagem e forma farmacêutica. Esse estudo pode ser considerado um indicativo de bioequivalência entre os medicamentos e, em alguns casos, pode ser utilizado em substituição ao estudo de bioequivalência.
Para registro de um medicamento genérico no mercado brasileiro, a comprovação de equivalência farmacêutica e bioequivalência em relação ao medicamento referência é um estudo necessário. Dessa forma, regularizar a forma que esse estudo é feito é de extrema importância, de maneira que fique claro como deve ser feito. Isso garante, ao final do estudo de equivalência e bioequivalência, a intercambiabildiade entre os medicamentos.
De acordo com o professor e especialista em Suporte Analítico da Brainfarma, Renato Cesar de Souza, há vários pontos importantes dessa RDC, já que ela não só guia o estudo de equivalência farmacêutica como também os indícios sobre procedimentos para desenvolvimento de método de perfil de dissolução, e apresenta o conceito de método de dissolução discriminativo, estudo de solubilidade e das variáveis a serem avaliadas.
“Existem alguns pontos importantes que precisam de um maior esclarecimento, como o critério adotado pela Agência Nacional de Vigilância Sanitária (Anvisa) para considerar a bioequivalência soberana em relação ao perfil de dissolução”, ressalta Souza.
Além disso, o método estatístico apresentado para comparação de perfis de dissolução é limitado para produtos que apresentam baixa variabilidade, não levando em consideração casos em que a forma farmacêutica apresenta uma variabilidade de liberação in vitro intrínseca. “Claro que existem ferramentas para produtos com liberação de alta variabilidade, mas, elas estão em fase de discussão e definição sobre a melhor aplicabilidade ao setor farmacêutico”, defende Souza.
Segue abaixo a RDC 31/10, com todos os seus artigos na íntegra. Para facilitar o entendimento do leitor, nós estamos dispondo todas as explicações no decorrer do texto da norma, em negrito, logo após cada artigo em questão. Os comentários em todos os artigos foram feitos pelo professor Renato Cesar de Souza.
RESOLUÇÃO-RDC Nº 31, DE 11 DE AGOSTO DE 2010
Dispõe sobre a realização dos Estudos de Equivalência Farmacêutica e de Perfil de Dissolução Comparativo.
A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária (Anvisa), no uso da atribuição que lhe confere o inciso IV do art. 11 do Regulamento aprovado pelo Decreto Nº 3.029, de 16 de abril de 1999, e tendo em vista o disposto no inciso II e nos §§ 1º e 3º do art. 54 do Regimento Interno aprovado nos termos do Anexo I da Portaria Nº 354 da Anvisa, de 11 de agosto de 2006, republicada no DOU de 21 de agosto de 2006, em reunião realizada em 5 de agosto de 2010, adota a seguinte Resolução e eu Diretor-Presidente determino a sua publicação:
CAPÍTULO I
DAS DISPOSIÇÕES PRELIMINARES
Art. 1º Esta Resolução dispõe sobre os requisitos para a realização dos Estudos de Equivalência Farmacêutica e de Perfil de Dissolução Comparativo, a serem atendidos pelos Centros de Equivalência Farmacêutica e Patrocinador do Estudo.
Essa RDC 31/10 surgiu para substituir a RE 310, de 01 de setembro de 2004. Trata-se de uma resolução mais completa e detalhada, comparada com a anterior. O objetivo principal dessa resolução é o de minimizar as dúvidas dos Patrocinadores e Centros de Equivalência Farmacêutica sobre os testes necessários para garantir a equivalência farmacêutica entre os produtos e um maior detalhamento sobe o teste de Perfil de Dissolução Comparativo. A resolução traz um texto bastante didático, e até exemplificativo, mas com alguns pontos que geram dúvida e diferentes interpretações.
Art. 2º Definições:
I - Acessório: complemento destinado a dosar, conduzir ou executar a administração da forma farmacêutica ao paciente. Comercializado dentro da embalagem secundária, junto com o medicamento e sem o contato direto com a forma farmacêutica;
II - Alta Solubilidade: é considerada altamente solúvel a substância ativa cuja quantidade correspondente a sua maior dose posológica disponível no mercado nacional é solúvel em 250mL ou menos de meio aquoso em uma escala de pH de 1,2-6,8 em uma temperatura de 37 ± 1ºC;
O conceito de alta solubilidade é baseado no sistema de classificação biofarmacêutico publicado por Amidon em 1995. Vale destacar que, nesse caso, a dose que deve ser levada em consideração é a maior dose posológica disponível no mercado nacional, ou seja, se há a possibilidade em bula de ser administrada duas ou mais doses do medicamento de uma vez, deve-se levar em consideração a soma dessas tomadas para verificar se a substância ativa é de alta solubilidade ou não.
III - Centro de Equivalência Farmacêutica: laboratório habilitado pela Anvisa que realiza os ensaios físico-químicos mínimos e, quando aplicáveis, microbiológicos ou biológicos mínimos dos Estudos de Equivalência Farmacêutica e de Perfil de Dissolução Comparativo, de pelo menos uma das formas farmacêuticas: sólidas, líquidas e semi-sólidas, responsabilizando-se técnica e juridicamente pela veracidade dos dados e informações constantes dos estudos, nos termos desta Resolução, sem prejuízo das atribuições do Patrocinador do Estudo;
Os estudos de equivalência farmacêutica e perfis de dissolução devem ser realizados por laboratórios, públicos ou privados, cadastradados, habilitados e supervisionados pela Anvisa, denominados Centros de Equivalência Farmacêutica, que pertencem à Rede Brasileira de Laboratórios Analíticos em Saúde (Reblas).
IV - Centro Responsável pelo Estudo: centro contratado pelo Patrocinador do Estudo, responsável pelos Estudos de Equivalência Farmacêutica e de Perfil de Dissolução Comparativo;
O patrocinador do estudo é qualquer empresa que tenha o interesse de comprovar a equivalência in vitro do medicamento desenvolvido em relação a um medicamento referência, e o Centro Responsável pelo Estudo é o laboratório habilitado pela Anvisa escolhido pelo patrocinador, que garante por meios de ensaios in vitro a equivalência entre os medicamentos.
V - Certificado de Equivalência Farmacêutica: documento elaborado pelo Centro de Equivalência Farmacêutica que atesta os resultados e conclui sobre o Estudo de Equivalência Farmacêutica, excluindo os dados brutos;
O Certificado de Equivalência Farmacêutica é o documento que aprova os medicamentos como equivalentes farmacêuticos. Trata-se de um documento necessário para submissão de processos de registro ou pós-registro perante a Anvisa, em que se garante por meio de um conjunto de ensaios físico-químicos e, quando aplicável, microbiológicos e biológicos, que os dois medicamentos são equivalentes.
VI - Certificado de Perfil de Dissolução Comparativo: documento elaborado pelo Centro de Equivalência Farmacêutica que atesta os resultados e conclui sobre o Estudo de Perfil de Dissolução Comparativo, excluindo os dados brutos;
O teste de Perfil de Dissolução é um teste físico-químico importante para demonstrar in vitro o desempenho de produtos que necessitam de dissolução para absorção e, consequentemente, efeito terapêutico.
O ensaio de perfil de dissolução é normalmente aplicado a formas farmacêuticas sólidas de administração por via oral, mas não restrito a essas formas. Para a equivalência farmacêutica dessas formas farmacêuticas sólidas de administração oral onde a dissolução se aplica como teste de verificação de qualidade do produto, há a necessidade, além da emissão do certificado de equivalência farmacêutica, da emissão do Certificado de Perfil de Dissolução, em que, por meio desse certificado, tem-se uma comprovação in vitro de que o medicamento teste tem o mesmo desempenho de liberação in vitro do medicamento referência.
VII - Dados Brutos: todos os registros e evidências que resultam de observações originais e das atividades de um determinado estudo. Podem incluir registros de dados, tabelas, cromatogramas, espectros, fotografias, dados manuscritos, dados eletrônicos, entre outros;
VIII - Dissolução muito rápida: dissolução média de no mínimo 85% da substância ativa em até 15 minutos;
IX - Dissolução rápida: dissolução média de no mínimo 85% da substância ativa em até 30 minutos;
Conceito muito importante para o perfil de dissolução, em que a velocidade de dissolução do produto de liberação imediata passa a ser dividida em duas classes, rápida ou muito rápida. Esse conceito é de extrema importância, principalmente na fase de comparação de perfis de dissolução.
X - Ensaios Informativos: ensaios analíticos preconizados na monografia individual ou nos métodos gerais de compêndios oficiais ou, ainda, em normas e regulamentos aprovados/referendados pela Anvisa, para os quais não exista especificação definida, cujos resultados não devem ser utilizados para fins de comparação entre os Medicamentos Teste e de Referência/Comparador no Estudo de Equivalência Farmacêutica. Para tais ensaios, o medicamento teste deve cumprir com suas próprias especificações;
Ensaios informativos são normalmente utilizados com o objetivo de caracterizar o produto desenvolvido, um exemplo claro de um ensaio informativo é a avaliação de peso médio de um comprimido de medicamento genérico. O medicamento referência não precisa atender a mesma especificação do medicamento teste, devido às possíveis diferenças em relação à massa do comprimido, visto que os processos podem ser diferentes. Todavia, o medicamento teste tem uma especificação a ser atendida a fim de garantir a qualidade do produto.
XI - Estudo de Equivalência Farmacêutica: conjunto de ensaios físico-químicos e, quando aplicáveis, microbiológicos e biológicos, que comprovam que dois medicamentos são Equivalentes Farmacêuticos;
Com o objetivo de garantir, que um medicamento tem a mesma eficácia e segurança de outro medicamento, o Estudo de Equivalência é uma parte dos estudos necessários. Esse estudo garante in vitro a semelhança entre os medicamentos, mas ela somente pode não ser suficiente para garantir que ambos têm a mesma segurança, eficácia e qualidade.
Testes como o de bioequivalência servem para comprovar a equivalência entre os produtos in vivo. Os estudos de equivalência, e bioequivalência, quando necessários, são de alta relevância para a saúde pública e interesses socioeconômicos, pois são ferramentas que garantem a intercambiabilidade entre os medicamentos lançados no mercado.
XII - Estudo de Perfil de Dissolução Comparativo: ensaio analítico com coletas em múltiplos pontos para a avaliação da dissolução de uma determinada substância ativa comparando duas formulações;
O estudo de perfil de dissolução fornece uma informação importante sobre a característica de liberação do fármaco da forma farmacêutica. A coleta e doseamento das amostras em múltiplos pontos ocorrem em diferentes tempos, em um intervalo adequado para a caracterização da velocidade de dissolução do fármaco.
De forma resumida, o principal objetivo do perfil de dissolução comparativo é verificar se a velocidade de dissolução do medicamento teste é semelhante à velocidade de liberação do medicamento referência, visto que a velocidade de liberação pode estar correlacionada com a velocidade de absorção do medicamento in vivo. Essa é uma das poucas ferramentas in vitro capazes de dar uma estimativa do comportamento do medicamento in vivo. Entretanto, para essa ferramenta ter um poder adequado de caracterização da velocidade de liberação uma série de testes precisam ser realizados, a fim de garantir que a condição adotada é a mais adequada possível.
XIII - Equivalentes Farmacêuticos: são medicamentos que possuem mesma forma farmacêutica, mesma via de administração e mesma quantidade da mesma substância ativa, isto é, mesmo sal ou éster da molécula terapêutica, podendo ou não conter excipientes idênticos, desde que bem estabelecidos para a função destinada. Devem cumprir com os mesmos requisitos da monografia individual da Farmacopéia Brasileira, preferencialmente, ou com os de outros compêndios oficiais, normas ou regulamentos específicos aprovados/referendados pela Anvisa ou, na ausência desses, com outros padrões de qualidade e desempenho. Formas farmacêuticas de liberação modificada que requerem reservatório ou excesso podem conter ou não a mesma quantidade da substância ativa, desde que liberem quantidades idênticas da mesma substância ativa em um mesmo intervalo posológico;
Para a comparação entre um medicamento teste e um medicamento referência, o objetivo principal do Patrocinador do Estudo, ao contratar o Estudo de Equivalência Farmacêutica, é de comprovar a equivalência entre os dois medicamentos por meio dos testes físico-químicos.
De acordo com esta RDC, os métodos que garantem a semelhança entre ambos os medicamentos podem ser encontrados na Farmacopéia Brasileira, preferencialmente, ou em monografias de outros compêndios oficiais específicos aprovados pela Anvisa.
É possível verificar na Resolução 37, de 6 e julho de 2009, que trata da admissibilidade das farmacopeias estrangeiras, os compêndios oficiais aceitos pela Anvisa, os quais podem ser adotados como monografia oficial, dentre eles, as farmacopeias alemã, americana, francesa, argentina, japonesa, britânica, mexicana, europeia e portuguesa.
XIV - Forma Farmacêutica: estado final de apresentação que os princípios ativos farmacêuticos possuem, após uma ou mais operações farmacêuticas executadas com a adição de excipientes apropriados ou sem a adição de excipientes, a fim de facilitar a sua utilização e obter o efeito terapêutico desejado, com características apropriadas a uma determinada via de administração;
XV - Forma Farmacêutica de Liberação Imediata: forma farmacêutica em que a dose total da substância ativa é disponibilizada rapidamente após sua administração. Em ensaios in vitro apresenta, em geral, dissolução média de no mínimo 75% da substância ativa em até 45 minutos. Tal forma farmacêutica pode ainda apresentar tipos de dissoluções diferenciadas em rápida e muito rápida;
XVI - Forma Farmacêutica de Liberação Prolongada: forma farmacêutica que apresenta liberação modificada em que a substância ativa é disponibilizada gradualmente da forma farmacêutica por um período de tempo prolongado;
XVII - Forma Farmacêutica de Liberação Retardada: forma farmacêutica que apresenta liberação modificada em que a substância ativa é liberada em um tempo diferente daquele imediatamente após a sua administração. As preparações gastro-resistentes são consideradas forma de liberação retardada, pois são destinadas a resistir ao fluido gástrico e liberar a substância ativa no fluido intestinal;
A norma apresenta uma definição clara dos principais tipos de forma farmacêutica sólida de administração por via oral.
XVIII - Medicamento Comparador: medicamento submetido ao Estudo de Perfil de Dissolução Comparativo para fins de mudanças pós-registro de medicamentos, conforme legislação específica, com o qual o Medicamento Teste será comparado;
Nem sempre a comparação em um estudo de equivalência se resume a um medicamento referência e um medicamento genérico. Em alguns casos de mudança pós-registro, não há a necessidade de um estudo de bioequivalência para garantir que a alteração proposta afeta a biodisponibilidade do produto. Um perfil de dissolução comparativo entre a condição aprovada e a proposta é suficiente. Nesses casos, o medicamento Comparador é o produto da condição aprovada, e o produto proveniente da condição proposta é o medicamento Teste.
XIX - Medicamento de Referência: medicamento inovador registrado no órgão federal responsável pela vigilância sanitária e comercializado no País, cuja eficácia, segurança e qualidade foram comprovadas cientificamente junto ao órgão federal competente, por ocasião do registro;
A lista de medicamentos referência da Anvisa, disponível em http://portal.anvisa.gov.br/registros-e-autorizacoes/medicamentos/produtos/medicamentos-de-referencia/lista, permite verificar todos os medicamentos referência comercializados no País.
Os medicamentos são divididos em Lista A e B, em que a lista A contém os medicamentos com apenas um insumo farmacêutico, e a Lista B contém os medicamentos com dois ou mais insumos farmacêuticos ativos em uma única forma farmacêutica. A verificação constante dessa lista é necessária devido às atualizações de medicamentos na lista de referência.
XX - Medicamento Teste: medicamento submetido aos Estudos de Equivalência Farmacêutica e de Perfil de Dissolução Comparativo;
XXI - Método de Dissolução Discriminativo: método capaz de evidenciar mudanças significativas nas formulações e nos processos de fabricação dos medicamentos testados que podem afetar o desempenho da formulação;
Talvez este seja um dos pontos de maior discussão atualmente. Um método de dissolução adequado deve ter uma capacidade discriminativa adequada. Isso porque, se existe a possibilidade de correlação entre velocidade de dissolução e velocidade de absorção do fármaco - e o método de dissolução tem capacidade de identificar alterações na velocidade de dissolução provenientes de alterações de processo - esse método passa a ter capacidade de minimizar os riscos aos pacientes de receber um medicamento com eficácia diferente daquele no momento de lançamento. O conceito parece adequado, mas nem sempre o desenvolvimento de um método com essa característica é simples.
XXII - Patrocinador do Estudo: pessoa jurídica, pública ou privada, que apoia financeiramente os Estudos de Equivalência Farmacêutica e de Perfil de Dissolução Comparativo, co-responsável técnica e juridicamente, juntamente com o Centro Responsável pelo Estudo, pela veracidade dos dados e informações constantes dos estudos;
XXIII - Protocolo de Estudo de Equivalência Farmacêutica: documento elaborado pelo Centro de Equivalência Farmacêutica que detalha a maneira como será realizado o Estudo de Equivalência Farmacêutica;
XXIV - Protocolo de Estudo de Perfil de Dissolução Comparativo: documento elaborado pelo Centro de Equivalência Farmacêutica que detalha a maneira como será realizado o Estudo de Perfil de Dissolução Comparativo;
XXV - Protocolo de Validação Parcial de Métodos Analíticos: documento elaborado pelo Centro de Equivalência Farmacêutica que detalha a maneira como será realizada a Validação Parcial de Métodos Analíticos;
XXVI - Relatório de Estudo de Equivalência Farmacêutica: documento elaborado pelo Centro de Equivalência Farmacêutica que atesta os resultados e conclui sobre o Estudo de Equivalência Farmacêutica, incluindo os dados brutos;
XXVII - Relatório de Estudo de Perfil de Dissolução Comparativo: documento elaborado pelo Centro de Equivalência Farmacêutica que atesta os resultados e conclui sobre o Estudo de Perfil de Dissolução Comparativo, incluindo os dados brutos;
XXVIII - Relatório de Validação Parcial de Métodos Analíticos: documento elaborado pelo Centro de Equivalência Farmacêutica que atesta os resultados e conclui sobre a Validação Parcial de Métodos Analíticos, incluindo os dados brutos;
Esses são documentos emitidos pelo centro de equivalência após a conclusão do estudo, que podem ser divididos em dois grupos: Protocolo e Relatório. O Protocolo tem a finalidade de descrever, de forma detalhada, como o estudo será feito, e deve ser elaborado e aprovado antes da execução dos experimentos. Já o relatório apresenta os resultados e conclusões obtidas nos experimentos realizados.
XXIX - Substância Química de Referência Farmacopéica (SQR): substância ou mistura de substâncias estabelecidas e distribuídas por farmacopéias ou instituições públicas oficiais autorizadas, possuindo alto grau de pureza e uniformidade. São planejadas para uso em ensaios químicos e físicos, nos quais suas propriedades são comparadas com as dos produtos que estão sendo analisados;
XXX - Substância Química de Referência Caracterizada (SQT): material de referência não estabelecido por farmacopéias ou instituições públicas oficiais autorizadas, devendo possuir alto grau de pureza e uniformidade. Deve ser cuidadosamente analisada em sua identificação, caracterização, impurezas e análise quantitativa; e
XXXI - Validação Parcial de Método Analítico: avaliação de alguns parâmetros de validação de métodos analíticos, quando houver transferência de metodologia do Patrocinador do Estudo para o Centro de Equivalência Farmacêutica.
A validação parcial tem o objetivo de comprovar que o método analítico previamente validado tem as características necessárias para obtenção de resultados com a qualidade exigida, nas condições em que é praticado. Esse inciso foi revogado na publicação da RDC 166, de 24 de julho de 2017.
CAPÍTULO II
DO ESTUDO DE EQUIVALÊNCIA FARMACÊUTICA
Seção I
Das Considerações Gerais do Estudo de Equivalência Farmacêutica
Art.3º O Estudo de Equivalência Farmacêutica deve ser realizado:
I - por Centro de Equivalência Farmacêutica devidamente habilitado pela Anvisa para essa finalidade, previamente à realização do Estudo de Biodisponibilidade Relativa/Bioequivalência, quando aplicável à forma farmacêutica;
Os centros habilitados pela ANVISA podem ser encontrados em http://portal.anvisa.gov.br/centros-de-equivalencia-farmaceutica.
II - comparando, simultaneamente, Medicamento Teste e Medicamento de Referência;
Ao exigir a análise simultânea do medicamento teste e referência, entende-se que não é possível, por exemplo, utilizar um resultado de perfil de dissolução de um medicamento referência feito no passado com um resultado de perfil de dissolução de medicamento teste feito depois.
III - com lotes dentro do prazo de validade.
- §1º Os medicamentos já registrados na Anvisa devem estar acondicionados em suas embalagens comerciais.
- §2º No caso de realização de estudos com lotes-piloto, os medicamentos devem estar acondicionados, no mínimo, em sua embalagem primária, devidamente identificada conforme legislação vigente, incluindo acessório, se aplicável.
O único caso em que não há necessidade do medicamento estar condicionado em suas embalagens comerciais é na realização de estudos com lote piloto, visto que, nessa fase piloto, a embalagem secundária do medicamento ainda não foi desenvolvida. Nesse caso, o medicamento em embalagem primária é suficiente, e nos casos em que há necessidade de um acessório para administração do medicamento, ele deve estar incluso.
- §3º O Estudo de Biodisponibilidade Relativa/Bioequivalência, a que se refere o inciso I, deve utilizar obrigatoriamente os mesmos lotes dos Medicamentos Teste e de Referência empregados no Estudo de Equivalência Farmacêutica.
O estudo de equivalência farmacêutica é uma forma de garantir que os medicamentos que serão utilizados no estudo de bioequivalência cumprem os requisitos de qualidade normatizados pela legislação vigente do País, minimizando os riscos aos indivíduos participantes do estudo.
Art. 4º O Estudo de Equivalência Farmacêutica pode ser realizado com medicamentos que se apresentem na forma de comprimido revestido/drágea, cujo Medicamento de Referência seja comprimido simples ou vice-versa, desde que o revestimento não controle a liberação da substância ativa.
Não há necessidade do medicamento teste apesentar o mesmo tipo de revestimento adotado pelo medicamento referência, desde que o revestimento do medicamento referência não tenha função de controle da liberação do fármaco.
Art. 5º No caso de formas farmacêuticas administradas como gotas, deve ser determinado o número de gotas que corresponde a 1mL, indicando-se a quantidade de substância ativa por gota.
Parágrafo único. A diferença permitida em relação ao número determinado de gotas por mililitro do Medicamento Teste é de até mais ou menos 10% em relação ao valor nominal declarado na bula do Medicamento de Referência.
Detalhes de como executar o teste de gotejamento para determinação do número de gotas por mililitro podem ser obtidos no capítulo geral da farmacopeia brasileira.
Art. 6º Não é aceito Estudo de Equivalência Farmacêutica realizado com Medicamentos Teste e de Referência acondicionados em embalagens primárias destinadas a dosar/conduzir/executar a administração de suas formas farmacêuticas ou que contenham acessórios que exijam ensaios específicos diferentes. Exemplo: solução oral que utiliza colher de medida, que não exige ensaio de gotejamento, não pode ser comparada com solução oral utilizando frasco gotejador, que exige o ensaio de gotejamento.
Art. 7º Para as formas farmacêuticas isentas do Estudo de Biodisponibilidade Relativa/Bioequivalência, conforme disposto em normas e regulamentos específicos aprovados/referendados pela Anvisa, a diferença de teor entre os Medicamentos Teste e de Referência pode ser superior a 5%, desde que ambos estejam dentro da especificação do método analítico adotado.
As formas farmacêuticas isentas do Estudo de Biodisponibilidade Relativa Bioequivalência estão disponíveis na Resolução nº 37, de 3 de agosto de 2011, e dispõe sobre o guia para isenção e substituição de estudos de biodisponibilidade relativa/bioequivalência e dá outras providências.
Art. 8º Para as formas farmacêuticas não-isentas do Estudo de Biodisponibilidade Relativa/Bioequivalência, recomenda-se que a diferença de teor da substância ativa entre os Medicamentos Teste e de Referência não seja superior a 5%.
A palavra recomenda-se nesse artigo não deixa um tom de obrigatoriedade de atendimento desse item, todavia, uma diferença de teor superior a 5% entre medicamento Teste e Referência pode ter impacto significativo no estudo de bioequivalência/biodisponibilidade relativa.
Seção II
Dos Critérios para a Realização do Estudo de Equivalência Farmacêutica
Art. 9º Os Medicamentos Teste e de Referência devem cumprir, em sua totalidade, com os requisitos da monografia individual da Farmacopéia Brasileira, preferencialmente, ou com os de outros compêndios oficiais, normas ou regulamentos específicos aprovados/referendados pela Anvisa, quando aplicáveis, complementados com os ensaios descritos em métodos gerais da Farmacopéia Brasileira e de outros compêndios oficiais para a forma farmacêutica em estudo.
A farmacopeia tem um papel fundamental na sociedade, considerada o Código Oficial Farmacêutico do País. A publicação estabelece os requisitos mínimos de qualidade para fármacos, insumos, drogas vegetais, medicamentos e produtos para a saúde.
Art.10 Na ausência de monografia descrita em compêndio oficial, normas ou regulamentos específicos aprovados/referendados pela Anvisa, deve-se utilizar método analítico validado pelo Patrocinador do Estudo ou Centro de Equivalência Farmacêutica.
Nem todos os medicamentos dispõem de monografias descritas em compêndios oficiais. Isso requer que o fabricante do genérico estabeleça os testes a serem realizados e valide os métodos analíticos a serem utilizados, ao transferir os métodos são convalidados posteriormente pelo EQFAR.
- §1º No caso citado no caput desse artigo, o Estudo de Equivalência Farmacêutica deve ser complementado com os ensaios descritos em métodos gerais da Farmacopéia Brasileira e de outros compêndios oficiais, normas ou regulamentos específicos aprovados/referendados pela Anvisa, para a forma farmacêutica em estudo.
No caso de um desenvolvimento de métodos devido à ausência de monografia descrita em compêndios oficiais, o Patrocinador do estudo deve, além dos métodos desenvolvidos, aplicar os métodos gerais da Farmacopéia, que são aplicáveis ao produto que está em fase de desenvolvimento.
- §2º Os Medicamentos Teste e de Referência devem cumprir com as mesmas especificações e os resultados dos ensaios não informativos do Medicamento Teste devem ser comparativos aos do Medicamento de Referência.
O método desenvolvido pelo Patrocinador deve ser capaz de caracterizar tanto o medicamento teste como o medicamento referência, pois o mesmo será utilizado para comprovar a equivalência entre os dois produtos.
Art.11. Quando o método analítico for transferido pelo Patrocinador do Estudo, o Centro de Equivalência Farmacêutica deve realizar a validação parcial desse método, previamente ao Estudo de Equivalência Farmacêutica.
Parágrafo único. A validação parcial deve cumprir com os requisitos dispostos no anexo I desta Resolução e seus parâmetros devem observar as normas e regulamentos específicos aprovados/referendados pela Anvisa.
O parágrafo único do artigo 11 e o anexo 1 foram revogados com a publicação de RDC 166, de 24 de julho de 2017. Nessa nova resolução, a validação parcial deve avaliar, pelo menos, os parâmetros de precisão, exatidão e seletividade. Nos casos de métodos analíticos destinados à quantificação de impurezas, a validação parcial deve incluir o limite de quantificação.
Art. 12. Não é aceito Estudo de Equivalência Farmacêutica em que se utilizem métodos e especificações de monografias de diversos compêndios oficiais para um mesmo estudo.
Parágrafo único. Quando a Farmacopéia Brasileira ou outro compêndio oficial apresenta monografia para determinado medicamento em que não estão contemplados todos os ensaios necessários para a comprovação de equivalência farmacêutica, o estudo deve ser complementado por ensaios de outro compêndio oficial, normas ou regulamentos aprovados/referendados pela Anvisa ou de outros padrões de qualidade aplicáveis utilizando método validado.
Utilizar um método de teor descrito na farmacopeia britânica e um método de dissolução da farmacopeia brasileira para avaliar a equivalência entre os produtos é um exemplo do que não é permitido de acordo com a resolução. Não pode haver troca de metodologia, porém pode haver complementação de ensaios, diante da ausência do ensaio na monografia adotada para avaliação da equivalência entre os medicamentos.
Art.13. São considerados ensaios informativos para fins de equivalência farmacêutica:
I - aspecto;
II - viscosidade;
III - densidade;
IV - valor do peso médio; ou
V - valor do volume médio.
- §1º As variações do peso médio e do volume médio para cada medicamento testado não são informativas e as especificações farmacopéicas devem ser cumpridas.
- §2º Os ensaios mencionados nesse artigo não são tratados como informativos quando forem importantes para a determinação da qualidade, segurança e eficácia dos medicamentos ou apresentarem especificações descritas em compêndios oficiais, normas ou regulamentos aprovados/referendados pela Anvisa.
Importante destacar que o valor do peso médio ou volume podem ser considerados como testes informativos, porém a faixa de variação desses itens deve atender às especificações descritas em farmacopeia. A farmacopeia brasileira descreve nos métodos gerais, como executar o teste de peso médio de acordo com a forma farmacêutica e as especificações a serem adotadas.
Art. 14. Na ausência de método de dissolução descrito em compêndio oficial, normas ou regulamentos específicos aprovados/referendados pela Anvisa, é de responsabilidade do Patrocinador do Estudo o relatório de desenvolvimento e validação do método de dissolução que deve ser realizado conforme preconizado em guias nacionais e internacionais e conter dados que demonstrem que o método é discriminativo.
I - o Centro Responsável pelo Estudo deve arquivar cópia do relatório de desenvolvimento do método de dissolução fornecido pelo Patrocinador do Estudo;
II - o Centro Responsável pelo Estudo deve proceder à validação parcial do método de dissolução desenvolvido e transferido pelo Patrocinador do Estudo; e
III - o relatório de desenvolvimento de dissolução deve conter, no mínimo, as seguintes informações:
- a) avaliação quantitativa da solubilidade da substância ativa na faixa de pH fisiológico (1,2 a 6,8), considerando a temperatura de 37°C ± 1°C, conforme, por exemplo, o método de diagrama de fase para análise de solubilidade. A avaliação requer que quantidades crescentes da substância ativa sejam testadas em volume fixo de, pelo menos, três diferentes meios como, por exemplo, em pH 1,2; 4,5 e 6,8;
- b) demonstração de que o meio de dissolução é o mais adequado à substância ativa na forma farmacêutica em estudo. A demonstração requer a investigação de curvas de dissolução na faixa de pH fisiológico (1,2 a 6,8), como, por exemplo, em pH 1,2; 4,5 e 6,8, considerando a temperatura de 37°C ± 1°C;
- c) demonstração de que o aparato, a rotação e os filtros utilizados no procedimento de coleta de amostras são os mais adequados à substância ativa e à forma farmacêutica em estudo;
- d) justificativa da necessidade da utilização de âncoras, quando aplicável;
- e) comprovação da necessidade de uso de tensoativos, bem como da quantidade empregada, quando aplicável;
- f) demonstração e justificativa da escolha do valor de Q (quantidade de substância ativa dissolvida expressa como porcentagem do valor rotulado da dose unitária); e
- g) justificativa da necessidade da aplicação de método de deaeração, quando aplicável.
- §1º O relatório de desenvolvimento do método de dissolução também pode ser adotado quando o método de dissolução descrito em compêndio oficial, normas ou regulamentos específicos aprovados/referendados pela Anvisa, não é adequado para o produto, desde que devidamente comprovado.
- §2º O pH do meio de dissolução deve contemplar a faixa fisiológica (1,2 a 6,8). Caso seja necessária a utilização de outra faixa de pH, essa deve ser justificada no relatório de desenvolvimento do método de dissolução.
- §3º O Patrocinador do Estudo pode contratar Centro de Equivalência Farmacêutica habilitado pela Anvisa para o desenvolvimento e validação do método de dissolução.
O Art. 14 apresenta uma descrição dos requisitos mínimos a serem avaliados para o desenvolvimento do método de dissolução, caso não exista um descrito em compêndio oficial ou caso o método descrito em compêndio oficial não seja adequado para o produto.
Com o objetivo de complementar esse artigo da resolução, a Anvisa publicou a Nota técnica nº 03, de 2013, que dispõe sobre a avaliação da solubilidade de fármacos e o desenvolvimento de métodos de dissolução para estudos de equivalência farmacêutica e perfil de dissolução comparativo, e dá outras providências.
Também, em maio de 20018, foi publicada a proposta do Guia de Dissolução Aplicável a Medicamentos Genéricos, Novos e Similares. As publicações desse guia e da nota técnica permitem esclarecer uma série de dúvidas sobre o que é um método de dissolução adequado e o que seria um método de dissolução descrito em compêndio oficial, que não é adequado para o produto. Fica claro que o objetivo principal de um método de dissolução é sua capacidade discriminativa, seja o método farmacopeico ou não.
O conceito de método de dissolução discriminativo ainda é muito discutido. Uma das grandes dificuldades consiste em obter um método discriminativo para formas farmacêuticas de liberação imediata de fármacos classificados como de alta solubilidade. No guia publicado em 2018, a agência reguladora propõe um método de dissolução padrão com especificação definida para fármacos de alta solubilidade, de acordo com o sistema de classificação biofarmacêutico.
Outro ponto importante é quais parâmetros do processo produtivo devem ser alterados e qual a faixa máxima permitida para comprovar que o método de dissolução tem capacidade discriminativa adequada. Esse item exige da equipe que desenvolve o método de dissolução um conhecimento profundo dos parâmetros críticos do processo farmacêutico e dos atributos críticos de qualidade do produto. Deve ser apresentado um racional adequado para justificar as alterações propostas e as faixas a serem avaliadas, de forma a comprovar que o método de dissolução proposto tem capacidade de identificar diferenças significativas do processo produtivo que podem ou não afetar a biodisponibilidade do produto.
Art. 15. O Estudo de Equivalência Farmacêutica de sprays e aerossóis nasais e pulmonares deve ser realizado conforme compêndios oficiais, normas ou regulamentos específicos aprovados/referendados pela Anvisa.
Art.16. Para sprays e aerossóis administrados por via não contemplada no artigo 15, devem ser realizados os ensaios farmacopéicos para a forma farmacêutica em questão. Exemplo: para solução spray administrado por via dermatológica devem ser realizados todos os ensaios da monografia individual e métodos gerais preconizados para a forma farmacêutica solução.
Parágrafo único. Quando os medicamentos citados no caput desse artigo possuírem dose definida em sua posologia, também deve ser comprovada a concentração da substância ativa por dose.
Devido à crescente demanda de registro, pós-registro e renovação de registros de produtos dessa classe a Anvisa publicou a Nota Técnica 001/2013, que dispõe sobre o ensaio e seus respectivos procedimentos para condução de estudo de equivalência farmacêutica de spray nasal, aerossóis nasais, MDIs orais e DPIs orais, bem como das análises estatísticas de bioequivalência populacional aplicáveis a determinados ensaios.
CAPÍTULO III
DO ESTUDO DE PERFIL DE DISSOLUÇÃO COMPARATIVO
Seção I
Das Considerações Gerais do Estudo de Perfil de Dissolução Comparativo
Art. 17. O Estudo de Perfil de Dissolução Comparativo deve ser realizado:
I - por Centro de Equivalência Farmacêutica devidamente habilitado pela Anvisa para essa finalidade, previamente ao Estudo de Biodisponibilidade Relativa/Bioequivalência, quando aplicável;
II - utilizando o mesmo método de dissolução empregado no Estudo de Equivalência Farmacêutica, quando aplicável;
III - utilizando os mesmos lotes dos Medicamentos Teste e de Referência empregados nos Estudos de Equivalência Farmacêutica e de Biodisponibilidade Relativa/Bioequivalência, quando aplicáveis;
IV - simultaneamente entre Medicamento Teste e Medicamento de Referência/Comparador; e
V - com lotes dentro do prazo de validade.
- §1º Os medicamentos já registrados na Anvisa devem estar acondicionados em suas embalagens comerciais.
- §2º No caso de realização de estudos com lotes-piloto, os medicamentos devem estar acondicionados, no mínimo, em sua embalagem primária, devidamente identificada conforme legislação vigente.
- §3º Nos casos de pós-registro, em que o Estudo de Equivalência Farmacêutica não é aplicável, o Estudo de Perfil de Dissolução Comparativo deve ser realizado utilizando método de dissolução descrito na Farmacopéia Brasileira, preferencialmente, ou em outros compêndios oficiais, normas ou regulamentos específicos aprovados/referendados pela Anvisa. Na ausência de monografia publicada em compêndio oficial, normas ou regulamentos específicos aprovados/referendados pela Anvisa, proceder conforme os critérios do artigo 14 desta Resolução.
As mesmas recomendações para um estudo de equivalência farmacêutica se aplicam para o Estudo de Perfil de Dissolução Comparativo.
Art. 18. Para formas farmacêuticas de liberação prolongada, a coleta de amostra deve ser representativa do processo de dissolução em, por exemplo, 1, 2 e 4 horas e depois a cada duas horas até que ambos os medicamento apresentem dissolução de 80% da substância ativa ou o platô seja alcançado.
Nas formas farmacêuticas de liberação prolongada, a substância ativa é disponibilizada gradualmente da forma farmacêutica por um período de tempo prolongado. Dessa forma, é necessária uma frequência adequada da amostragem. Os tempos iniciais do perfil de dissolução são úteis para identificar possíveis eventos de liberação muito rápida da forma farmacêutica e o final da dissolução serve para garantir a liberação total do ativo.
Importante destacar que não é obrigatório atingir pelo menos 80% de liberação ao final do teste. Em alguns casos, o platô, que é um indicativo de interrupção do processo de dissolução, é suficiente para caracterizar a dissolução do produto.
Art. 19. Para formas farmacêuticas de liberação retardada deve ser realizada dissolução em meio HCl 0,1N durante 2 horas (etapa ácida), seguida de dissolução em meio tampão. Após o momento em que se coloca o medicamento no meio tampão, a coleta de amostra deve ser representativa do processo de dissolução em, por exemplo, 15, 30, 45, 60 e 120 minutos até que ambos os medicamentos apresentem dissolução de 80% da substância ativa ou o platô seja alcançado.
As formas farmacêuticas de liberação retardada mais conhecida são as preparações gastro-resistentes, em que a forma farmacêutica é destinada a resistir ao fluido gástrico e liberar a substância ativa no fluido intestinal. Nesses casos, a etapa ácida sozinha não caracteriza o perfil de dissolução do produto, mas a sua resistência gástrica. Após a etapa ácida, a velocidade de liberação do fármaco é avaliada em uma condição em que a dissolução do produto deve ocorrer. O início da dissolução na etapa em tampão tende a ser mais lento que uma forma farmacêutica de liberação imediata, por isso os primeiros tempos de coleta tendem ser maiores.
Art. 20. O Estudo de Perfil de Dissolução Comparativo pode ser realizado com medicamentos que se apresentem na forma de comprimido revestido/drágea, cujo Medicamento de Referência/Comparador seja comprimido simples ou vice-versa, desde que o revestimento não controle o mecanismo de liberação da substância ativa.
Art. 21. Quando o resultado do Estudo de Perfil de Dissolução Comparativo for não semelhante, a comprovação da equivalência terapêutica entre os Medicamentos Teste e de Referência/Comparador pode, a critério da ANVISA, ser baseada no resultado do Estudo de Biodisponibilidade Relativa/Bioequivalência.
Mesmo diante de resultados de perfil de dissolução não semelhante, a agência reguladora pode considerar a bioequivalência soberana. Todavia a Anvisa não deixa claro, em nenhum momento, quais os critérios adotados para considerar a soberania da bioequivalência em relação a perfil de dissolução in vitro. A não semelhança de perfil de dissolução, mas com resultado positivo no estudo de bioequivalência, pode ocorrer por vários motivos, um deles pode estar relacionado justamente com as técnicas aceitas pelos órgãos regulatórios para comparação de métodos de perfis de dissolução.
O método adotado pela Anvisa é o de modelos independentes de fator de similaridade (f2) e classificação de perfis de dissolução como rápido ou muito rápido. Esses métodos são mundialmente aceitos e são boas ferramentas para classificar um perfil como semelhante ou não, devido à simplicidade envolvida nos cálculos e os critérios que são de fácil compreensão.
Entretanto, essas diferenças podem não refletir as diferenças encontradas in vivo, por exemplo, diferenças pequenas entre a classificação de perfil de dissolução, como rápido ou muito rápido, podem não implicar necessariamente que o resultado in vivo reflita essa diferença. A decisão de verificar se essa diferença é crítica para a performance do produto no teste do estudo de bioequivalência cabe à empresa, que assume o risco de ter um resultado reprovado.
Além disso, como o desenvolvimento de método de dissolução tem o objetivo principal de caracterizar o mecanismo de liberação da formulação teste, e esse método deve ter capacidade discriminativa adequada para essa formulação, não é incomum obter um método de dissolução que tenha capacidade discriminativa suficiente para detectar diferença entre o perfil de dissolução do medicamento teste e medicamento referência, mas diferenças que podem não ter impacto in vivo. Nesses casos, o risco de decidir enviar para o estudo de bioequivalência, mesmo diante da não semelhança entre perfis de dissolução, continua sendo da empresa e, ao obter uma bioequivalência positiva, o risco ao paciente é minimizado visto que existe uma possibilidade do método de dissolução desenvolvido ter um poder discriminativo excessivo.
Considerar a bioequivalência soberana pode implicar no risco de a agência reguladora entender que, para a comprovação da adequação de todas as futuras mudanças pós-registro do produto, a empresa deverá conduzir novo estudo de biodisponibilidade relativa, afinal, no momento em que, por exemplo, o perfil de dissolução comparativo não comprova semelhança, perde-se a base para comparações posteriores.
Portanto, todas as futuras alterações pós-registro do produto irão se fundamentar na comprovação de bioequivalência, cujo estudo teria que ser analisado previamente pela Anvisa para o deferimento do pleito, impossibilitando, portanto, a implementação imediata da alteração por parte da empresa.
Art. 22. Não se aplica a realização do Estudo de Perfil de Dissolução Comparativo para as seguintes formas farmacêuticas:
I - pós, granulados e formas farmacêuticas efervescentes que ao serem reconstituídos tornam-se soluções;
II - semi-sólidos, excetuando-se supositórios;
III - formas farmacêuticas administradas como sprays ou aerossóis nasais ou pulmonares de liberação imediata;
IV - gases; ou
V - líquidos, exceto suspensões.
- §1º Para as formas farmacêuticas citadas, quando houver metodologia de dissolução descrita em compêndio oficial, normas ou regulamentos específicos aprovados/referendados pela Anvisa, o Estudo de Perfil de Dissolução Comparativo, ou ensaio complementar a critério da Anvisa, deve ser realizado.
- §2º Para as formas farmacêuticas não citadas, deve ser realizado o Estudo de Perfil de Dissolução Comparativo.
Em algumas formas farmacêuticas o teste de perfil de dissolução não se aplica, normalmente por não ser necessária a dissolução para a ação farmacêutica do fármaco, como, por exemplo, as soluções em que o fármaco já se encontra dissolvido.
Seção II
Da Comparação de Perfis de Dissolução
Art. 23. A comparação de perfis de dissolução é útil nos casos em que se deseja conhecer o comportamento de dois medicamentos antes de submetê-los a Estudo de Biodisponibilidade Relativa/Bioequivalência, para isenção de menores dosagens desses estudos e para alterações pós-registro.
Art. 24. Nesta comparação avalia-se a curva como um todo empregando o Método Modelo Independente Simples.
I - um Método Modelo Independente Simples é aquele que emprega um fator de diferença (F1) e um fator de semelhança (F2). Nos termos desta Resolução, os perfis de dissolução comparativos são avaliados apenas utilizando-se o cálculo do fator de semelhança (F2); e
II - o fator F2 corresponde a uma medida de semelhança entre as porcentagens dissolvidas de ambos os perfis:
onde: n = número de tempos de coleta considerados para fins de cálculo de F2; Rt = valor de porcentagem dissolvida no tempo t, obtido com o Medicamento de Referência ou Comparador; Tt = valor de porcentagem dissolvida do Medicamento Teste ou da formulação alterada, no tempo t.
Parágrafo único. O fator de semelhança (F2) somente deve ser calculado quando as condições do ensaio de dissolução forem exatamente as mesmas empregadas na avaliação dos Medicamentos Teste e de Referência/Comparador.
O único método aceito em legislação pela Anvisa é o método de modelo independente de fator de similaridade (f2). Trata-se de um método simples e adequado na maioria das vezes, mas apresenta algumas limitações.
Subseção I
Do Procedimento para Comparação de Perfis de Dissolução
Art. 25. A comparação de perfis de dissolução deve seguir os seguintes procedimentos:
I - empregar doze unidades do Medicamento Teste e doze unidades do Medicamento de Referência/Comparador; e
O método de fator de similaridade se baseia na diferença entre a média de cada ponto de coleta, portanto, para se ter uma estimativa adequada da média é necessário um número de amostras adequado.
II - calcular o fator F2 utilizando a equação apresentada no inciso II do Art. 24.
Art. 26. Para que dois perfis de dissolução sejam considerados semelhantes, devem atender aos seguintes critérios:
I - os Medicamentos Teste e de Referência/Comparador devem apresentar tipos de dissoluções correspondentes. Por exemplo, se o Medicamento de Referência/Comparador apresentar dissolução média de 85% em 30 minutos (dissolução rápida) o Medicamento Teste deve apresentar também dissolução rápida;
II - o valor do fator de semelhança (F2) deve estar compreendido entre 50 a 100;
Um valor de F2 maior que 50 significa uma diferença média entre os pontos menor ou igual a 10%.
III - os tempos de coleta devem ser os mesmos para as duas formulações;
III - o número de pontos de coleta deve ser representativo do processo de dissolução até que se obtenha platô na curva, sendo obrigatória a quantificação de amostras de, no mínimo, cinco tempos de coleta;
IV - para fins de cálculo F2, utilizar, no mínimo, os três primeiros pontos, excluindo o tempo zero;
Para caracterizar um perfil de dissolução do produto é necessário uma amostragem de pelo menos cinco pontos, mas não significa que os cinco pontos serão utilizados para comparação pelo método de fator de similaridade, já que o mínimo necessário são três pontos.
V - para fins de cálculo F2, incluir apenas um ponto da curva após ambos os medicamentos atingirem a média de 85% de dissolução; e
A partir do momento em que o perfil de dissolução atinge 85%, o poder do teste de fator de similaridade em detectar diferenças diminui, visto que aumenta a possibilidade de se atingir um platô na curva de dissolução após esse ponto e os resultados serem próximos, tanto para o medicamento teste como para o de referência.
VI - para permitir o uso de médias, os coeficientes de variação para os primeiros pontos de coleta não podem exceder 20%. Para os demais pontos considera-se o máximo de 10%. São considerados como primeiros pontos de coleta o correspondente a 40% do total de pontos coletados. Por exemplo, para um perfil de dissolução com cinco tempos de coleta, consideram-se primeiros pontos os dois primeiros tempos de coleta.
Uma variabilidade alta implica em uma estimativa inadequada da média e, consequentemente, uma maior incerteza no valor de f2 obtido, já que o método não leva em consideração os valores individuas, mas somente a média de cada ponto de coleta. Por isso, o órgão regulador restringe o máximo de variabilidade permitida para alguns pontos. Todavia, a resolução não leva em consideração os produtos farmacêuticos que tem propriedade intrínseca de alta variabilidade no perfil de dissolução in vitro.
Existem algumas ferramentas reconhecidas em órgão internacionais como o EMA e FDA como o método de bootstrap e o método de modelo dependente de distância multivariada de Mahalanobis. Apesar de os guias internacionais citarem a possiblidade de uso dessas ferramentas, verifica-se muita discussão na comunidade científica sobre as vantagens e desvantagens dessas ferramentas. Por exemplo, apesar de o guia do EMA descrever a possibilidade do uso do método de modelo dependente de distância multivariada de Mahalanobis, a agência publicou, em agosto de 2018, um esclarecimento sobre o uso da ferramenta, recomendando aplicar o método de Bootstrap.
No Brasil, as discussões sobre a possibilidade de uso de ferramentas alternativas estão em andamento, mas, por enquanto, o único método reconhecido em legislação é o de fator de similaridade. Diante da impossibilidade de diminuição da variabilidade do perfil de dissolução in vitro - e comprovada a característica inerente dessa variabilidade relacionado ao produto - a empresa pode optar por discutir a proposta junto a Anvisa por meio de reuniões presenciais ou dos canais de comunicação.
Parágrafo único. Quando a substância ativa apresentar alta solubilidade e a formulação for de liberação imediata, apresentando dissolução muito rápida para ambos os medicamentos, o fator F2 perde o seu poder discriminativo e, portanto, não é necessário calculá- lo. Nesses casos deve-se comprovar a dissolução muito rápida dos produtos, por meio do gráfico da curva, realizando coletas em, por exemplo: 5, 10, 15, 20 e 30 minutos. O coeficiente de variação no ponto de 15 minutos que não pode exceder 10%.
Da mesma forma que os pontos após o platô diminuem o poder discriminativo do método de fator de similaridade, uma dissolução muito rápida dificulta a aplicação da ferramenta, pois selecionar três pontos representativos em um intervalo de 15 minutos pode ser inviável.
Um ponto importante nesse paragrafo único é que esse critério se aplica a substância ativa de alta solubilidade em formulação de liberação imediata. Parece que esse critério não leva em consideração casos de fármacos de baixa solubilidade que podem apresentar um método de perfil de dissolução rápido. Apesar de não ser um comportamento esperado para essa classe de fármacos, ao tentar mitigar a baixa solubilidade do fármaco adicionando-se normalmente surfactante ao meio de dissolução, a dissolução pode ser muito rápida e, mesmo assim, o método manter um poder discriminativo e uma especificação adequada. Ao limitar esse parágrafo a substâncias ativas de alta solubilidade, surgem dúvidas sobre o que fazer em casos de substâncias de baixa solubilidade.
CAPÍTULO IV
DAS AMOSTRAS PARA A REALIZAÇÃO DOS ESTUDOS DE EQUIVALÊNCIA FARMACÊUTICA E DE PERFIL DE DISSOLUÇÃO COMPARATIVO
Art. 27 A quantidade de amostras a ser adquirida pelo Centro deve possibilitar um Estudo completo de Equivalência Farmacêutica e de Perfil de Dissolução Comparativo e um reteste.
- §1º O prazo para a retenção dos lotes deve ser correspondente a, no mínimo, um ano após o prazo de validade do medicamento que expire por último.
- §2º Para as formas farmacêuticas estéreis, é obrigatória a realização dos ensaios de esterilidade e endotoxina bacteriana ou pirogênio no Estudo de Equivalência Farmacêutica, tanto para o Medicamento Teste como para o Medicamento de Referência/Comparador. As amostras de retenção referentes a esses ensaios são dispensadas para o Medicamento de Referência/Comparador.
CAPÍTULO V
DAS SUBSTÂNCIAS QUÍMICAS DE REFERÊNCIA PARA REALIZAÇÃO DOS ESTUDOS DE EQUIVALÊNCIA FARMACÊUTICA E DE PERFIL DE DISSOLUÇÃO COMPARATIVO
Art. 28. Deve-se utilizar Substância Química de Referência (SQR) oficializada pela Farmacopéia Brasileira, preferencialmente, ou por outros compêndios oficiais.
Art. 29. No caso da inexistência da SQR, será admitido o uso de Substância Química de Trabalho (SQT), desde que sejam devidamente determinados: identidade, teor, perfil quantitativo de impurezas e, quando aplicáveis, perfil qualitativo de impurezas e outros ensaios específicos.
- §1º É de responsabilidade do Patrocinador do Estudo ou do Centro Responsável pelo Estudo garantir a confiabilidade dos dados da SQT, por meio de uma análise crítica de seu laudo analítico.
- §2º O prazo de validade da SQT deve respeitar o prazo de validade da matéria-prima determinado por seu fabricante. Não são permitidas revalidações da matéria-prima pelo Patrocinador do Estudo e/ou Centro de Equivalência Farmacêutica, com o objetivo de extensão do prazo de validade da SQT.
O uso de substância química de referência de trabalho ou caracterizada no estudo de equivalência só é permitido diante a ausência dessas substâncias em órgãos oficiais reconhecidos pela Anvisa.
CAPÍTULO VIII
DOS CERTIFICADOS DOS ESTUDOS DE EQUIVALÊNCIA FARMACÊUTICA E DE PERFIL DE DISSOLUÇÃO COMPARATIVO
Art. 30. O Certificado do Estudo de Equivalência Farmacêutica deve obedecer aos seguintes critérios:
I - quando houver especificações quantificáveis, os resultados dos ensaios devem ser descritos como grandezas numéricas em unidades preconizadas pelos compêndios oficiais ou pelo Sistema Internacional de Medidas. Não são aceitos resultados descritos como "conforme", "de acordo" ou outros;
II - para o ensaio de esterilidade, admite-se a descrição dos resultados somente como: "estéril" ou "não estéril";
III - nos ensaios de dissolução, desintegração, peso médio, volume médio, dureza e uniformidade de doses unitárias devem ser informados: média, resultados mínimo e máximo e, quando aplicável, desvio padrão relativo/limite de variação, tanto para o Medicamento Teste quanto para o Medicamento de Referência/Comparador;
IV - nos resultados do ensaio de aspecto, devem ser descritas as características dos Medicamentos Teste e de Referência/Comparador, tais como: formato, dimensão, cor, presença de sulcos, presença de revestimento, gravações, odor característico ou outras que permitam identificar as amostras;
V - para metodologias descritas em compêndios oficiais, no campo "Referências Bibliográficas" do Certificado deve ser reportada a referência do compêndio adotado com, no mínimo, o ano, o fascículo, a edição e a página. Quando o compêndio utilizado for eletrônico, dispensa-se a informação do número da página; e
VI - para metodologias não descritas em compêndios oficiais, no campo "Referências Bibliográficas" do Certificado deve ser reportado o código de identificação da metodologia analítica adotada, bem como o código de identificação do respectivo Relatório de Validação.
O Artigo 30 apresenta as normas de como os resultados do estudo de equivalência devem ser apresentados. O principal objetivo é deixar claro que os resultados comparativos entre medicamento teste e medicamento referência/comparador devem ser reportados de forma clara, demonstrando a origem dos métodos analíticos utilizados e o valores obtidos para ambos os medicamentos nos testes realizados.
Art. 31. O Certificado do Estudo de Perfil de Dissolução Comparativo deve obedecer aos seguintes critérios:
I - no campo "Especificação do Método de Quantificação", além das especificações do método, deve ser reportado o critério de aceitação do ensaio;
II - para metodologias descritas em compêndios oficiais, no campo "Referências Bibliográficas" do Certificado deve ser reportada a referência do compêndio adotado com, no mínimo, o ano, o fascículo, a edição e a página. Quando o compêndio utilizado for eletrônico, dispensa-se a informação do número da página; e
III - para metodologias não descritas em compêndios oficiais, no campo "Referências Bibliográficas" do Certificado deve ser reportado o código de identificação da metodologia analítica adotada, bem como o código de identificação do respectivo Relatório de Validação.
CAPÍTULO IX
DAS DISPOSIÇÕES FINAIS
Art. 32. O Patrocinador do Estudo deve encaminhar à Anvisa o Certificado do Estudo de Equivalência Farmacêutica e de Perfil de Dissolução Comparativo, conforme modelos disponíveis no sítio eletrônico da Anvisa.
Há um modelo padronizado pela agência reguladora que se encontra disponível em http://portal.anvisa.gov.br/registros-e-autorizacoes/medicamentos/equivalencia-farmaceutica/certificados.
Art. 33. Devem estar à disposição da Anvisa e do Patrocinador do Estudo os Protocolos e Relatórios dos Estudos de Equivalência Farmacêutica, Perfil de Dissolução Comparativo, Validação e de Validação Parcial de Métodos Analíticos, bem como os dados brutos e estatísticos da avaliação de cada ensaio com os Medicamentos Teste e de Referência/Comparador.
Parágrafo único. É de responsabilidade do Centro Responsável pelo Estudo o arquivamento de toda a documentação citada no caput do artigo.
Art. 34. Documentação e ensaios adicionais podem ser solicitados a qualquer momento pela Anvisa para complementação da avaliação dos Estudos de Equivalência Farmacêutica, Perfil de Dissolução Comparativo, Validação e Validação Parcial de Métodos Analíticos.
Art. 35. Os Centros de Equivalência Farmacêutica devem observar as normas e regulamentos técnicos em vigor.
Art. 36. Esta Resolução entra em vigor após 60 dias de sua publicação oficial.
Art. 37. Fica revogada a Resolução-RE Nº 310, de 1º de setembro de 2004.
DIRCEU RAPOSO DE MELLO
ANEXO I
Esse anexo foi revogado com a publicação de RDC nº 166 de 2017 de 24 de julho de 2017.
REQUISITOS PARA A VALIDAÇÃO PARCIAL DE MÉTODOS ANALÍTICOS
- Os ensaios submetidos à validação parcial são classificados em quatro categorias segundo sua finalidade, conforme Tabela 1.
Tabela 1: classificação das categorias, segundo a finalidade dos ensaios:

- Para cada categoria de ensaio, a respectiva metodologia será considerada validada parcialmente, desde que avaliados o conjunto de parâmetros relacionados na Tabela 2.
Tabela 2: parâmetros necessários para a validação parcial do método analítico, segundo a categoria do ensaio:
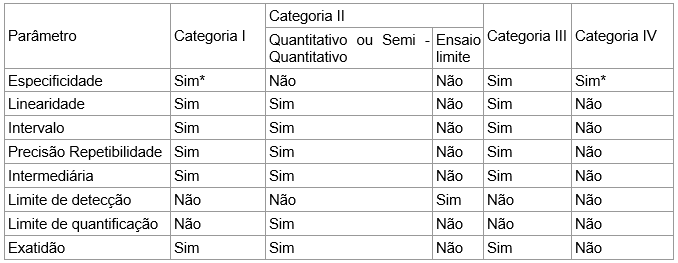
* O Centro deve solicitar o placebo ou adquirir cópia da documentação referente a esse parâmetro realizado pelo patrocinador na validação do método.

{kind=link}