





O desafio da implantação da Integridade de Dados nos processos e na Validação de Sistemas Computadorizados: as agências reguladoras do setor farmacêutico mundial de medicamentos sempre dependeram do conhecimento que as organizações desenvolveram para a fabricação, embalagem, análises, distribuição e monitoramento de produtos. Está implícita, no processo de avaliação e revisão, a confiança entre o órgão regulador e a indústria, que gera as informações enviadas em estudos, documentos de processo e liberações analíticas. Essas informações, que são a base das Boas Práticas de Fabricação (BPF) – ‘o que não foi escrito, não foi feito’ -, são usadas nas decisões diárias e devem, então, ser abrangentes, completas e confiáveis.
Essas decisões são tomadas com base em dados (em papel ou informatizados), que devem ser atribuíveis, legíveis, contemporâneos, originais, precisos, completos, consistentes, duradouros e disponíveis, bem como descrito no referido ALCOA+ e nas principais normativas nacionais e internacionais.
O documento Data Integrity and Compliance With CGMP Guidance for Industry, do Food and Drug Administration (FDA), que é a agência reguladora sanitária dos Estados Unidos, de 2016, menciona o termo ALCOA, para os itens aos quais um dados gerado deve atender, a sigla é composta então pelas iniciais, em inglês, dos itens a seguir:
Attributable: atribuíveis;
Legible: legíveis;
Contemporaneuous: contemporâneos;
Original: originais; e
Accurate: acurados.
A essa lista adicionamos os itens a seguir para atender à exigência do PIC/s, que atualmente é o conceito requerido para o Brasil, descritos, então, como ALCOA +:
Complete (Completo);
Consistent (Consistente);
Enduring (Duradouro); e
Available (Disponível).

Fig.: 1 – Demonstrativo do ALCOA +.
Os fatores contribuintes para que fosse dada tamanha importância e fizesse com que as indústrias farmacêuticas começassem a levar mais a sério o tema integridade de dados incluem falhas das organizações na aplicação de sistemas robustos que inibam os riscos aos dados gerados, para melhorar a detecção de situações em que a confiabilidade dos dados pode ser comprometida e para investigar e solucionar as causas principais quando ocorrem falhas e desvios. Essas observações destacam a necessidade da indústria de modernizar estratégias de controle histórico, aplicar o gerenciamento de riscos de qualidade, bem como princípios científicos sólidos e atualizar todo o processo de gerenciamento de dados.
Mesmo em países que são auditados pelo FDA, podemos encontrar um cenário que demonstra problemas, tal qual tem acontecido no Brasil, regulado pela Agência Nacional de Vigilância Sanitária (Anvisa). No caso do FDA, as atividades da indústria farmacêutica são regulamentadas pelo guia 21 CFR part 210 and 211. Na Europa, existe, também, um regulamento equivalente, incluído no Eudralex.
Cenário mundial
Falando um pouco sobre o cenário mundial, atualmente as indústrias farmacêuticas que cumprem a normativa do FDA necessitam de muita atenção para não entrarem na lista dos que possuem sistemas de qualidade farmacêuticos duvidosos e serem penalizados fortemente por isso. Essa avaliação envolve a fabricação de medicamentos, cosméticos e alimentos.
Um estudo realizado pelo FDA, entre 2015 e 2019, demonstrou que as infrações sanitárias estão presentes, em maior quantidade, nas empresas fabricantes de produtos acabados, quando comparados aos números obtidos nas inspeções a fabricantes de matérias primas. Entre as infrações estão incluídas as relacionadas à integridade de dados. Podemos visualizar na figura abaixo:
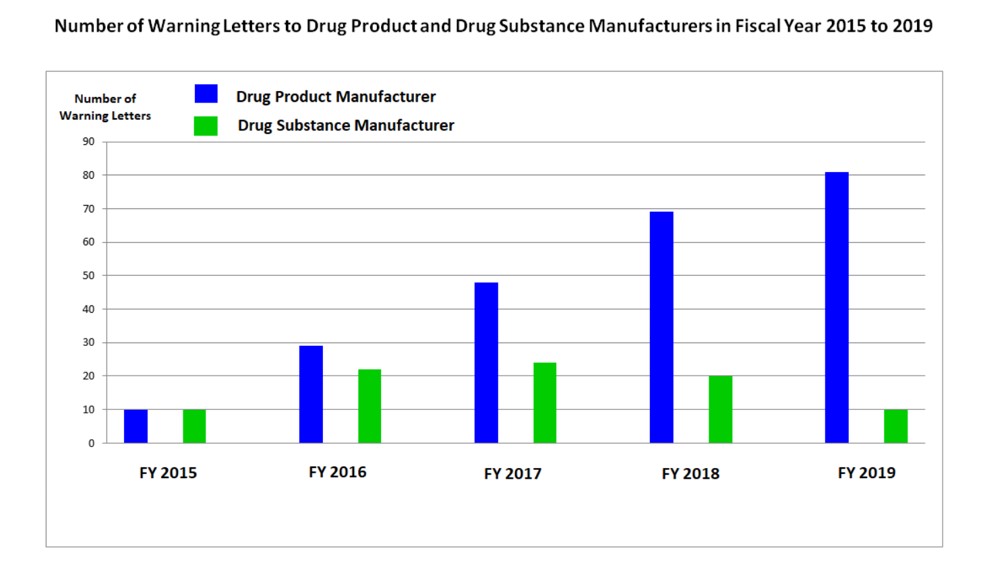
Fig.: 2 – Gráfico comparativo do número de relatórios com infrações apresentados pela indústria farmacêutica fabricante de produtos acabados (azul) e fabricantes de matérias primas (verde).
Em outro estudo foram avaliados o número de apontamentos por tema inspecionado pelo FDA. A partir dessa informação, foram ranqueados os cinco temas com o maior número de infrações sanitárias (que envolvem o recebimento de uma carta indicando o defeito encontrado, ‘warning letter’) nas indústrias farmacêuticas dos Estados Unidos, China, Índia e outras empresas do extremo oriente. São denominados, então, os ‘TOP FIVE’ das irregularidades.
A seguir, os temas avaliados, seguidos do item do guia do FDA, serão demonstrados em gráfico que ilustra a frequência relativa (em %) dos ‘TOP FIVE’ nos últimos cinco anos, compreendendo o período de outubro de 2014 a setembro de 2019:
- Boas Práticas de Fabricação nas áreas de responsabilidades da unidade de controle de qualidade (211.22);
- Procedimentos e desvios escritos (211.100);
- Teste e liberação para distribuição (211.165);
- Revisão de registros de produção (211.192);
Teste de estabilidade (211.166).
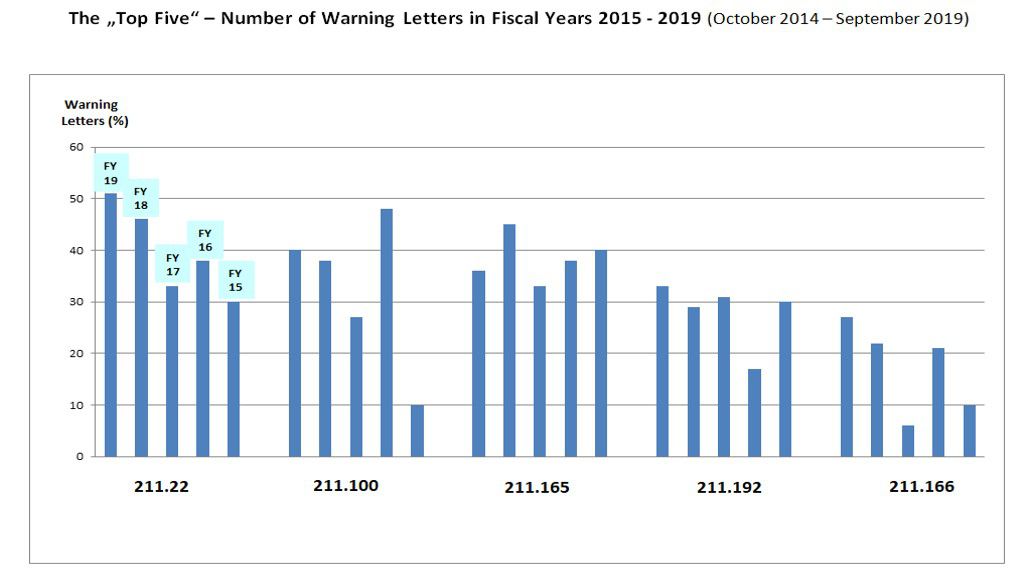
Fig.: 3 – ‘TOP FIVE’ no ranking do FDA para os temas com maior incidência de infrações sanitárias durante inspeções nas indústrias farmacêuticas.
Desafios mais críticos
Nas últimas décadas, uma estrutura bem estabelecida de gerenciamento da qualidade dos dados se tornou um dos desafios mais críticos para os fabricantes da indústria farmacêutica no mundo. Em paralelo a isso, notamos o aumento do número de observações feitas em relação às boas práticas de gerenciamento de dados durante as inspeções de BPF. As razões para esse aumento do nível de preocupação das autoridades de saúde com relação à confiabilidade dos dados são multifatoriais, e incluem maior conscientização regulatória e preocupação com a segurança dos controles apropriados e modernos.
Os fatores contribuintes incluem falhas das organizações na aplicação de sistemas robustos que inibem os riscos dos dados gerados, para melhorar a detecção de situações em que a confiabilidade dos dados pode ser comprometida e para investigar e solucionar as causas principais quando ocorrem falhas. Essas observações destacam a necessidade da indústria de modernizar estratégias de controle histórico, aplicar o gerenciamento moderno de riscos de qualidade, bem como princípios científicos sólidos e atualizar todo o processo de gerenciamento de dados.
No Brasil, a integridade dos dados também é obrigatória para o setor de saúde regulamentado, pois as decisões de processamento e disposição em relação à qualidade, segurança, eficácia, pureza e conformidade com os requisitos regulamentares aplicáveis são realizadas baseadas nos dados registrados e relatados.
É um dever da indústria gerar dados com precisão e consistência, que sejam armazenados seguramente e contem com a ausência de qualquer alteração desses dados, mesmo entre as atualizações de um registro de dados. É imposta dentro de um sistema em seu estágio de design, por meio do uso de regras e procedimentos padrão, e é mantida pelo uso de rotinas de verificação e validação de erros. É também um aspecto crítico para o design a implementação e uso de qualquer sistema computadorizado ou não que armazene, processe ou recupere dados.
Refere-se à manutenção e garantia da precisão e consistência dos dados durante todo o ciclo de vida.
Os dados são registrados exatamente como pretendidos e, após recuperação posterior, os dados são os mesmos de quando foram originalmente gravados, além de estarem completos, consistentes e precisos.
A RDC 301/19 e a IN 43/19 mencionam a importância da integridade de dados aplicados a sistemas informatizados ou não, incluindo também a verificação durante a validação de sistemas computadorizados, cada qual com suas particularidades, mas, dentro de um escopo lógico de atuação.
Esse assunto, em algumas indústrias farmacêuticas, ainda é um fator de grande dúvida e, para simplificar, demonstro abaixo um fluxo lógico que tem como objetivo avaliar o grau de conformidade do sistema em termos de integridade dos dados, com base em sua criticidade.
Esse processo é amplamente utilizado em países da Europa, Ásia e América Central, e está descrito na figura 4.
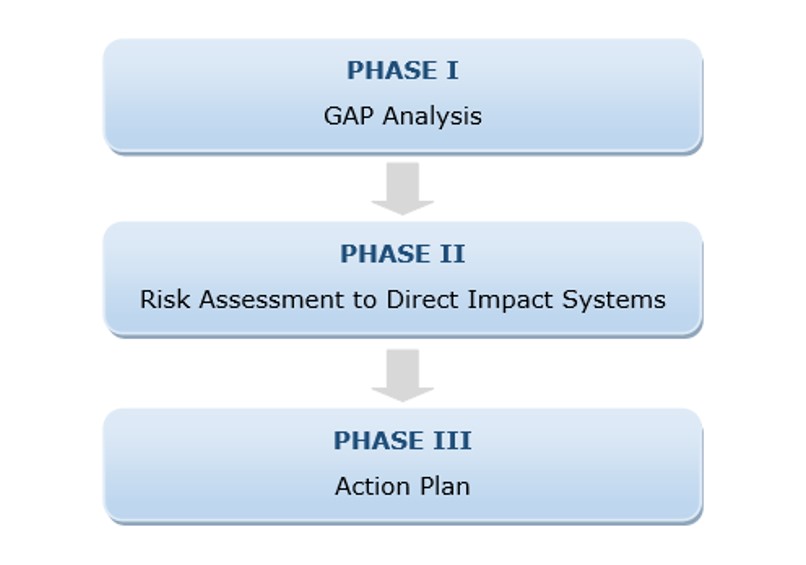
Fig.: 4 – Fases da Avaliação da Integridade de Dados.
Na Fase 1: Fazemos o estudo ‘GAP Analysis’, em que é realizada uma avaliação relacionada a todos os processos, procedimentos, equipamentos e sistemas (computadorizados ou não), que possam, de alguma maneira, influenciar na qualidade, segurança e eficácia dos produtos, ou que ainda possam afetar a saúde dos consumidores.
Essa avaliação inicial é um trabalho em conjunto da equipe de validação e das áreas que possuem relação com Boas Práticas, definindo o nível de confiabilidade e exposição dos dados gerados.
É avaliada a integridade, segurança e rastreabilidade dos dados emitidos, transmitidos e arquivados, juntamente com a conformidade dos sistemas computadorizados e as políticas de gerenciamento correspondentes com os requisitos das normas e padrões atuais de BPF. Entre esses padrões, destaca-se o mais conhecido por sistemas computadorizados, 21CFR parte11 da FDA
Os sistemas computadorizados são avaliados independentemente da geração de dados eletrônicos ou dados em papel, conforme descrito na figura 5.
São verificados todos os dados gerados, processados e armazenados, juntamente com as funcionalidades do sistema e também todos os procedimentos relacionados às práticas descritas aqui.
Com essas informações, os possíveis riscos serão identificados para serem, então, avaliados na próxima fase, a fase 2. O resultado da fase 1 é qualitativo, separando os sistemas, processos e procedimentos e equipamentos com impacto do restante, sem impacto ou que necessita de pequena remediação. Essa etapa tem o escopo de excluir de outras avaliações os sistemas com baixo impacto na qualidade do produto.
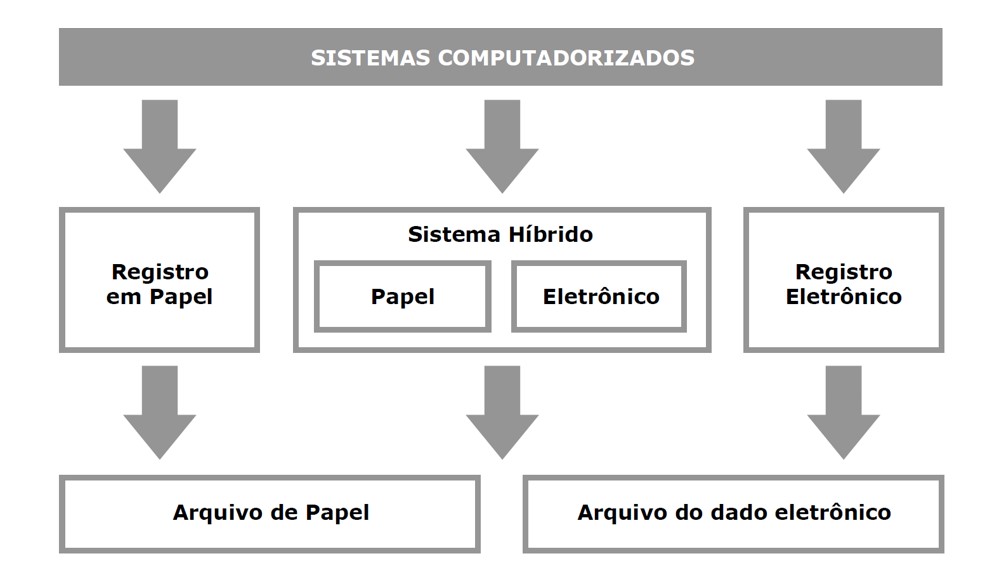
Fig.: 5 – Geração de dados por meio de Sistemas Computadorizados.
Fonte: Data Integrity in the FDA – Regulated Laboratory – April 2014 – tradução livre.
Na Fase 2 é realizada uma análise mais profunda. O impacto e a criticidade da qualidade induzidos por tecnologia complexa são avaliados e combinados. O resultado da análise agora é quantitativo, e um valor representando a pontuação do nível de risco é atribuído a cada cenário identificado. Isso ocorre, especificamente, nos dois aspectos (impacto na qualidade e criticidade da tecnologia), e são analisados individualmente. A avaliação inclui controles e medidas de gerenciamento de risco apropriados. O regulamento de referência para realizar essa avaliação de risco é o ICHQ9.
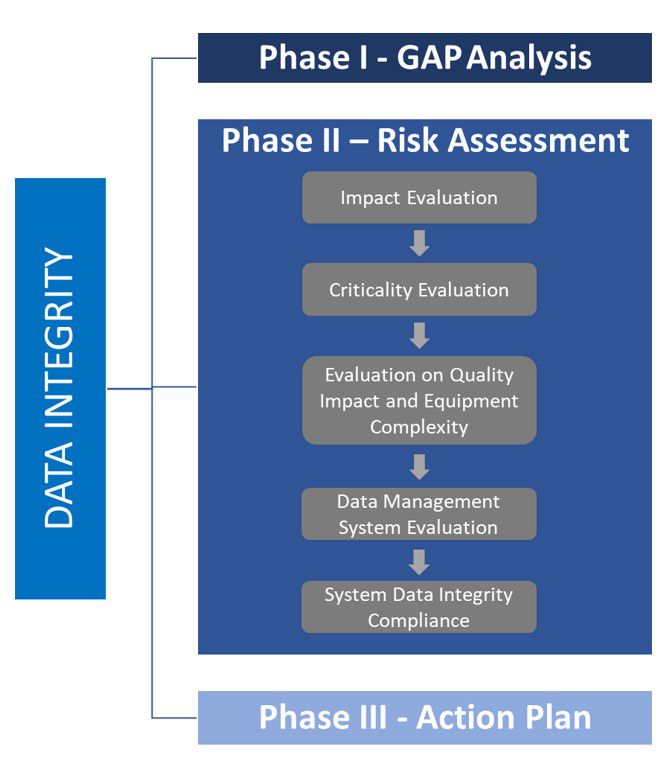
Fig.: 5 – Detalhamento da fase 2 sobre a Análise de Risco relacionada à Integridade de Dados.
Na Fase 3 é elaborado um Plano de Ação, ou também chamado de Plano de Remediação, voltado para a aplicação da ferramenta CAPA (Ação Corretiva e Ação Preventiva). Uma sugestão importante é que o plano de ação contenha algumas informações fundamentais, mencionadas abaixo:
- Item avaliado;
- Risco avaliado;
- Descritivo da ação proposta (preventiva ou corretiva);
- Área Responsável pela Ação;
- Pessoa responsável pela Ação;
- Prazo para a ação ser executada e estar concluída;
- Prazo de verificação da ação e informação de reprogramação, caso haja a possiblidade;
- Campo para anotações; e
- Verificação e aprovação do fechamento das ações.
É recomendado o uso de planilhas para a execução das atividades de Análise de Risco, o que agiliza todo o processo. Para isso, só devemos ter a preocupação com a validação das mesmas, caso exista essa indicação.
O assunto Integridade de Dados no Brasil ainda requer muita atenção de quem está começando, dos que estão em situação irregular conscientemente ou mesmo para quem quer se aprofundar no tema e trabalhar na área, pois a indústria farmacêutica brasileira tem sofrido duras penas no processo de adequação. Quanto antes as empresas se adequarem menor o risco de recall ou interdições graves ocorreram.
No campo profissional, existe ainda um pequeno e seleto grupo de profissionais que sabe como tratar, verdadeiramente, o assunto, bem como a exigência do mercado e a Anvisa o requer. É preciso buscar mais pela informação. A contratação de profissionais que não sabem o que fazer pode ser muito nociva para as empresas, o que, além de gastar muito tempo e dinheiro, têm a sensação de estar cobertas, quando, na verdade, essa ilusão se acaba durante a inspeção sanitária de uma forma muito triste.
A política do Brasil mudou, a regulação mudou, a ANVISA também mudou e as interdições têm sido cada vez mais uma realidade para quem não muda com a cultura de qualidade.
Agradecimentos: Carlos Martins (Azbil Telstar Portugal) e Rafael Beaus – (Azbil Telstar Espanha).
*FERNANDA BIDO é farmacêutica, professora do ICTQ – Instituto de Pesquisa e Pós-Graduação para o Mercado Farmacêutico e gerente de Consultoria na Azbil Telstar Brasil.
Abaixo, seguem algumas literaturas de pesquisa para quem gostaria de se envolver mais neste tema:
Referências Bibliográficas:
- ANVISA - RDC 301/2019 – Boas Práticas de Fabricação de Medicamentos;
- ANVISA – IN 43/2019 – Boas Práticas de Fabricação complementares
- aos sistemas computadorizados utilizados na fabricação de Medicamentos.
- Guia SINDUSFARMA de Integridade de Dados – 2017;
- TÍTULO 21, Código de Regulamentos Federais (21 CFR), Partes 210 e 211;
- Diretrizes de Boas Práticas de Fabricação (BPF) do EUDRALEX Volume 4 Anexo 11 - Sistemas Computadorizados;
- Diretrizes de Boas Práticas de Fabricação (BPF) EUDRALEX Volume 4 Anexo 15 - Qualificação e Validação;
- FDA 21 CFR Part 210 - Boas práticas atuais de fabricação na fabricação, processamento, embalagem ou retenção de medicamentos; geral;
- FDA 21 CFR Part 211 - Boas práticas atuais de fabricação de produtos farmacêuticos acabados;
- FDA 21 CFR Parte 11 - Registros Eletrônicos; Assinaturas eletrônicas;
- ISPE GAMP 5 - Uma abordagem baseada em risco para sistemas computadorizados GxP em conformidade;
- ICH Q9 - Gerenciamento de Riscos da Qualidade;
- Princípios Gerais de Validação de Software; Orientação Final para Indústria e Funcionários da FDA, janeiro de 2002;
- Orientação para a parte 11 do setor, registros eletrônicos, assinatura eletrônica - Escopo e Aplicação, agosto de 2003;
- Guia ISPE GAMP - Integridade de Registros e Dados;
- Definições e diretrizes de integridade de dados MHRA GMP para o setor, março de 2015;
- Preguntas e respostas da EMA: Boas práticas de fabricação. Integridade de dados;
- Orientação sobre boas práticas de gerenciamento de dados e registros. Série de relatórios técnicos da OMS nº 996 (2016);
- Boas práticas para gerenciamento e integridade de dados em ambientes regulamentados de BPF / PIB. PIC / S PI 041-1 (rascunho 2);
Participe também: Grupo de WhatsApp para receber notícias farmacêuticas diariamente



