A farmacodinâmica é o estudo do mecanismo de ação dos fármacos em geral, enquanto que a farmacocinética avalia os efeitos que o corpo faz com o fármaco, dentre eles, os processos de absorção, distribuição, metabolismo e excreção.
Segundo o farmacêutico e professor do ICTQ – Instituto de Pesquisa e Pós-Graduação para o Mercado Farmacêutico, Alexandre Massao Sugawara, até mesmo a mais promissora das drogas fracassará em seus intuitos terapêuticos se não alcançar concentrações mínimas efetivas no órgão alvo para exercer seus efeitos terapêuticos.
Numerosos fatores podem impedir este intento. Tais fatores são organizados em basicamente quatro etapas farmacocinéticas: absorção, distribuição, metabolismo e excreção. A depender das características físico-químicas de cada fármaco e da forma como interage com estruturas corpóreas este trajeto será facilitado ou dificultado. “Por isso, esse conhecimento é fundamental para a compreensão dos fatores que levam um fármaco a ser efetivo ou não”, ressalta Sugawara.
De modo geral, as interações dinâmicas entre a absorção, a distribuição, o metabolismo e a excreção de um fármaco determinam a sua concentração plasmática e estabelecem a capacidade do fármaco de alcançar o seu órgão-alvo numa concentração efetiva. Variações individuais, como perfis farmacogenéticos diversos, condições patológicas, idade, interações medicamentosas, podem modificar os parâmetros farmacocinéticos e alterar a efetividade de um medicamento. “É fundamental reconhecer como estas diversas variáveis podem modificar o perfil farmacocinético e a farmacodinâmica de um medicamento. Intervir, quando necessário, garante a plena efetividade terapêutica dos medicamentos”, fala o professor.
A farmacocinética clínica é uma ferramenta importante para auxiliar no alcance da posologia ideal, além de possibilitar a compreensão da ocorrência de variações entre grupos. O principal objetivo é garantir que as doses administradas sigam dentro de uma margem terapêutica que se sabe que é eficaz e segura para aquele doente. Este processo é apoiado em princípios farmacocinéticos que promovem uma utilização mais racional do medicamento.
Absorção dos fármacos
Sugawara lembra que, para que um fármaco seja absorvido pelo corpo e possa se distribuir pelos líquidos corporais, é preciso transpor barreiras teciduais que são compostas, em última análise, por membranas celulares que apresentam natureza lipoproteica. Assim, um composto químico, para se difundir através dessas membranas, dependerá de suas propriedades físico-químicas de miscibilidade em um meio predominantemente oleoso.
O coeficiente de partição octanol/água (P) indica a tendência de um composto em se difundir em óleo ou água. O logaritmo deste coeficiente é o LogP. Se P = 1, LogP = 0, indicando que a molécula apresenta a mesma afinidade por óleo e pela água. LogP > 0, indica que a molécula é lipofílica e se LogP < 0 indica que o composto é hidrofílico. Valores muito baixos de LogP dificultam a permeação pelas membranas celulares, por outro lado, valores muito elevados podem manter as moléculas retidas na membrana, devido a sua alta lipossolubilidade. O valor ideal para fármacos fica entre 2 a 5.
“Por mais estranho que possa parecer, uma molécula precisa ter, como vimos, certa lipossolubilidade para atravessar as barreiras naturais do corpo humano e também apresentar certa hidrossolubilidade, afinal nosso corpo é 70% água e a distribuição pelos líquidos corporais exige do fármaco propriedades de miscibilidade com a água”, explica o professor.
Se, por um lado, necessita de caráter lipofílico para permeação através de membranas celulares, por outro lado, se faz necessário ser hidrofílico para se distribuir pelo plasma. A principal estratégia da química farmacêutica para que um fármaco tenha as duas propriedades é criar compostos ionizáveis a diferentes faixas de pH.
Fármacos ácidos fracos, como a aspirina, em um ambiente altamente ácido, como o suco gástrico, predominam em sua forma protonada, não ionizada, lipossolúvel, tendendo a atravessar membranas biológicas da parede do estômago com mais facilidade. Ao alcançar o ambiente mais alcalino do plasma, a aspirina é desprotonada, tornando-se ionizada, aumentando sua hidrossolubilidade e dissolução no plasma, evitando sua reabsorção retrógrada.
Equação de Henderson-Hasselbalch
Estas propriedades físico-químicas das moléculas podem ser expressas matematicamente em pKa, que representa o grau que um fármaco estará num dos lados da membrana. A Equação de Henderson-Hasselbalch descreve a relação entre a pKa de um fármaco ácido ou básico e o pH do meio biológico.
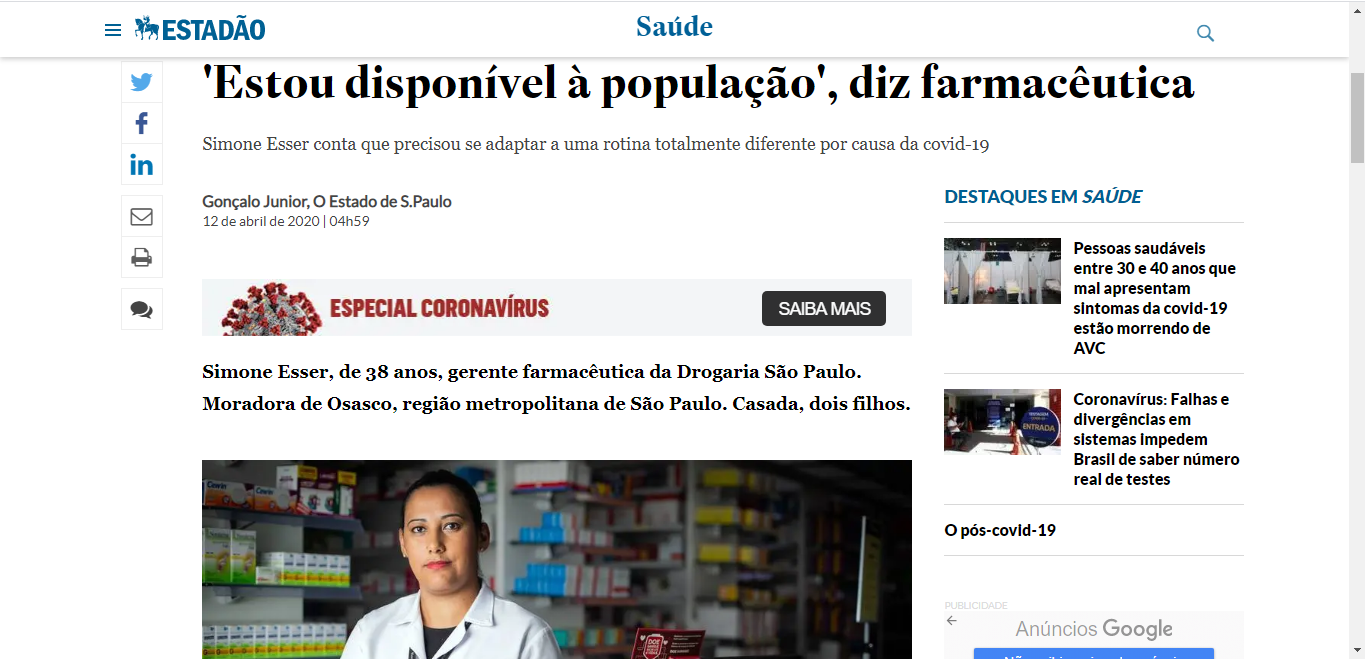 Considerando o pH do estômago como 1,0 e um fármaco ácido fraco com pKa 6,0 temos:
Considerando o pH do estômago como 1,0 e um fármaco ácido fraco com pKa 6,0 temos:
Como se vê, um fármaco ácido fraco com pKa 6,0 em meio ácido estará numa razão de 100.000 para 1 em sua forma protonada, não ionizada, lipossolúvel.
Agora, quando este fármaco alcança o plasma com pH 7,4, as condições do meio mudam. Assumiremos um pH do plasma 7,0 para exemplificar:
 “Como se vê, um fármaco ácido fraco com pKa 6,0 em meio alcalino, como o plasma, estará numa razão de 1 para 10 em sua forma desprotonada, ionizada, hidrossolúvel”, afirma o professor.
“Como se vê, um fármaco ácido fraco com pKa 6,0 em meio alcalino, como o plasma, estará numa razão de 1 para 10 em sua forma desprotonada, ionizada, hidrossolúvel”, afirma o professor.
Há, por certo, uma pequena quantidade de fármacos que não é planejada para se difundir através de membranas por simples difusão, mas utiliza-se de transportadores proteicos, e assim alcança o plasma. Os principais exemplos são fármacos peptídicos e aminoácidos que usam os mesmos transportadores utilizados pelos seus análogos nutricionais. Como exemplo, há a levodopa, um medicamento antiparkinsoniano.
Sugawara afirma que, atualmente estudam-se superfamílias de transportadores de membrana, como a ATP binding cassete (ABC), Solute Carrier (SLC) e Organic Solute Carrier (SLCO), encontradas nas mais diversas barreiras biológicas, como intestino, fígado, barreira hematoencefálica, placenta, glândulas mamárias e rins.
Por exemplo, o paracetamol é um substrato para a proteína transportadora ABCB1 e, ao mesmo tempo, um inibidor ABCB1, dificultando a absorção de fármacos transportados por esta família de transportadores. Uma fonte de consulta que o professor sugere é o DrugBank© (https://www.drugbank.ca), um banco de dados canadense de acesso livre sobre fármacos e medicamentos que dá informações neste sentido.
Há, ainda, a glicoproteína-P (P-gP) um transportador de efluxo presente em inúmeras membranas biológicas, como o intestino e a barreira hematoencefálica. Muitos autores indicam um papel de proteção da P-gP contra o acúmulo de xenobióticos no ambiente intracelular, eliminado esses compostos, por exemplo, na luz intestinal ou para fora do sistema nervoso central.
A cafeína é um inibidor da P-gP e seu uso concomitante com fármacos substratos da P-gP aumenta a concentração extra membrana desses compostos. “Características genéticas individuais para a expressão aumentada do P-gP podem explicar a não absorção de certos fármacos substratos da P-gP, como a digoxina”, lembra ele.
Vias de administração
A via de administração deve ser considerada na absorção do fármaco. Por exemplo, a insulina é uma molécula proteica e seria digerida antes de ser absorvida, portanto, sua via de administração é injetável. O professor destaca que a via parenteral é vantajosa devido a alcançar rapidamente picos plasmáticos em comparação com a via oral. Por outro lado, a via oral é de fácil aderência do paciente a esquemas terapêuticos e de baixo custo operacional. Há ainda as vias transdérmicas e inalatórias indicadas, sobretudo, para doenças que afetam a pele e o sistema respiratório, respectivamente, devido à baixa absorção sistêmica.
Um importante conceito que integra a absorção dos fármacos é a biodisponibilidade, que indica a fração absorvida do fármaco, resultado da via de administração, de propriedades físico-químicas do fármaco e de certos fatores presentes nos pacientes, como ocorre com os transportadores. É apresentada sob a fórmula:
 Na circulação, os fármacos podem sofrer a interferência da ligação com proteínas plasmáticas, uma forte tendência de se ligar com a albumina, nossa principal proteína plasmática, o que diminui a sua concentração no órgão-alvo. Como o fármaco apresenta uma distribuição limitada, seu volume de distribuição é normalmente baixo.
Na circulação, os fármacos podem sofrer a interferência da ligação com proteínas plasmáticas, uma forte tendência de se ligar com a albumina, nossa principal proteína plasmática, o que diminui a sua concentração no órgão-alvo. Como o fármaco apresenta uma distribuição limitada, seu volume de distribuição é normalmente baixo.
“A coadministração de fármacos transportados por proteínas plasmáticas levaria a um aumento de sua fração livre de menor afinidade, predispondo a indesejáveis efeitos tóxicos. Entretanto, são poucas as demonstrações desses tipos de interações na prática clínica”, lembra Sugawara.
Reações de metabolização
Após a fase distributiva, começam a se impor a metabolização dos fármacos que, a partir de um pico plasmático, a curva de concentração plasmática pelo tempo começa a declinar. As reações de metabolização são classificadas em dois tipos: de oxidação/redução e as de conjugação/hidrólise.
As reações de oxidação/redução modificam a estrutura química de um fármaco por meio de oxidação ou redução. O sistema do citocromo P450 reúne estas famílias de enzimas no fígado.
As reações de conjugação/hidrólise hidrolisam ou conjugam o fármaco com uma molécula grande e polar para inativar aquela substância ou, mais comumente, para aumentar a sua solubilidade e excreção na urina ou na bile. Em certas ocasiões, a hidrólise ou a conjugação pode resultar em ativação metabólica de pró-fármacos. Os grupos mais comumente adicionados incluem glicuronato, sulfato, glutationa e acetato.
Os efeitos das reações de oxidação/redução e de conjugação/hidrólise sobre determinado fármaco também dependem da presença de outros fármacos tomados concomitantemente.
Certas classes, como os barbitúricos, são poderosos indutores de enzimas que medeiam reações de oxidação/redução. Outros fármacos são capazes de inibir essas enzimas. A compreensão dessas interações medicamentosas constitui um pré-requisito essencial para a dosagem apropriada de associações de fármacos.
“Atualmente, reconhece-se substratos, inibidores e indutores para cada subtipo de enzima da família do citocromo P450. Recomendo, mais uma vez, o DrugBank© (https://www.drugbank.ca), como fonte de informação e a Micromedez© acessada por meio do site www.psbe.ufrn.br”, sugere o professor.
Excreção
Após a fase de biotransformação, e mesmo em paralelo a ela, tem o início a excreção do fármaco, que pode ser modificada por fatores que mudam a filtração glomerular e a reabsorção e secreção tubular.
De acordo com o que Sugawara já afirmou, fatores que afetam a taxa de filtração glomerular (TFG) também são capazes de influenciar a taxa de depuração da droga. A inflamação dos capilares glomerulares aumenta a pressão hidrostática capilar e aumenta a filtração das drogas.
Como proteínas não são filtráveis pelo glomérulo, as drogas que exprimem certa afinidade com proteínas plasmáticas teriam suas taxas de filtração diminuídas. A faixa habitual de meias-vidas observada para a maioria das drogas exclusivamente depuradas por filtração glomerular é de uma a quatro horas. Entretanto, serão observadas meias-vidas consideravelmente mais longas, caso ocorra grande ligação a proteínas.
Após a filtração pelo glomérulo, é iniciado o processamento tubular do filtrado em dois processos: reabsorção e secreção tubular. A reabsorção de fármacos através dos túbulos depende do pH intraluminal. Como o pH do filtrado é ácido, isso favorece a reabsorção de fármacos ácidos de volta ao corpo e, pelo mesmo motivo, um fármaco básico seria facilmente excretado. Por exemplo, a alcalinização da urina é uma estratégia para sequestrar a aspirina e mantê-la no interior dos túbulos e, assim eliminá-la do corpo nos casos de overdose.
Transportadores
O professor explica, ainda, que alguns fármacos sofrem secreção direta para dentro dos túbulos renais, que podem conter mecanismos de transporte renal de ânions e cátions orgânicos. Os transportadores de ânions orgânicos permitem a secreção de muitos ânions orgânicos, entre eles os antibióticos (cefalosporinas e penicilinas), contrastes radiológicos, diuréticos (furosemida), analgésicos e seus metabólitos, conjugados ou não.
Este transporte pode ser tão eficiente que alguns ânions são quase removidos completamente durante a primeira passagem pelo rim. A probenecida e o lítio podem inibir o sistema de secreção tubular de ânions orgânicos e reter ou diminuir a excreção de fármacos que se utilizam destes mecanismos.
O sistema de transporte catiônico é responsável pela secreção de bases fracas que estão ionizados no pH plasmático, como, por exemplo, a cimetidina, a metadona, a morfina e a procainamida. A P-glicoproteína é uma bomba de efluxo que também secreta múltiplos cátions orgânicos. A digoxina é um exemplo de fármaco secretado pela Pgp no túbulo proximal renal e este transporte pode ser inibido pela quinidina, verapamil e ciclosporina A, indicando o mecanismo envolvido nesta importante interação medicamentosa.
Clearance
A depuração ou clearance de um fármaco é o parâmetro que integra todos os fenômenos farmacocinéticos da metabolização e excreção de um fármaco, e indica mais significativamente o tempo de ação dele em seus alvos moleculares, celulares e orgânicos.
Ao diminuir a concentração do fármaco ativo no sangue, o metabolismo e a excreção reduzem o tempo durante o qual um fármaco é capaz de atuar sobre um órgão alvo. A meia-vida de eliminação é definida como o tempo durante o qual a concentração do fármaco no plasma diminui para a metade de seu valor original. O conhecimento da meia-vida de eliminação permite calcular a frequência de doses necessária para manter a sua concentração plasmática dentro da faixa terapêutica.
 “Fatores que afetam o volume de distribuição de fármaco modificam seu tempo de meia-vida. A diminuição da massa muscular, comum no envelhecimento, minimiza o volume de distribuição e, consequentemente, a meia-vida do fármaco. Já a obesidade, aumenta o volume de distribuição e a meia-vida do fármaco”, comenta Sugawara.
“Fatores que afetam o volume de distribuição de fármaco modificam seu tempo de meia-vida. A diminuição da massa muscular, comum no envelhecimento, minimiza o volume de distribuição e, consequentemente, a meia-vida do fármaco. Já a obesidade, aumenta o volume de distribuição e a meia-vida do fármaco”, comenta Sugawara.
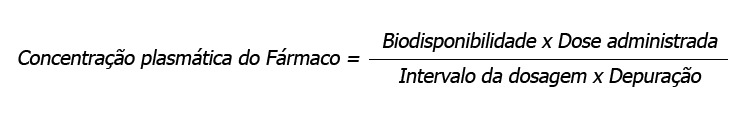
Tipicamente, os esquemas posológicos ótimos mantêm a concentração plasmática do fármaco no estado de equilíbrio dinâmico dentro de sua janela terapêutica. Como o estado de equilíbrio dinâmico é alcançado quando a taxa de aporte do fármaco é igual à sua eliminação, a concentração no estado de equilíbrio dinâmico é afetada pela razão entre o produto de sua biodisponibilidade e dose administrada pelo intervalo entre doses e sua depuração. Este ponto é alcançado em cerca de quatro a cinco meias-vidas.
“Por tudo isso, eu gostaria de enfatizar que é importante o farmacêutico ter pleno conhecimento prático sobre a farmacocinética clínica e farmacodinâmica. O farmacêutico deve exercer, de fato, seu papel social, atendendo à demanda de saúde da população. Não é mais concebível que um profissional de saúde, com tantas capacidades, permaneça subutilizado em farmácias, drogarias e hospitais. Façamos o seguinte, saia detrás do balcão da farmácia, levante-se de sua cadeira de escritório, vá ao encontro dessas necessidades e mostre a amplitude de suas competências”, finaliza o professor.